Getting Started
Get started by completing the total energy tutorial.
Input files
RESCU+ reads input files in the JSON format. The JSON format is lightweight, portable and essentially composed of Python’s basic types like: dict, list, int, float, str. It is thus convenient to prepare, read or write input files in Python using the JSON module. RESCU+ accepts so-called exhaustive input files, which contain almost all of a simulation parameters, including:
atomic orbital basis functions (if any)
geometry
pseudopotentials
solver parameters
etc.
Such files are convenient to keep as reference input, since they are not susceptible to changes in default parameters between version, among other advantages. But it is impractical to produce them from manually. Which is why RESCU+ works in tandem with nanotools, a Python interface which makes it convenient to create structures, write input files and read the output files produced by RESCU+.
System and calculator classes
Atomic systems are described by the System class. System objects
can be initialized from an Atoms and a Cell object (or their
corresponding dictionaries). System objects have additional
attributes (such as Kpoint) which are described
here. nanotools includes various
calculator classes like the basic (and most important) TotalEnergy
class. A TotalEnergy instance holds all the data regarding a given
calculation:
the system description and simulation parameters;
the solvers’ parameters;
the output data.
A TotalEnergy object can be initialized from a System object or
a corresponding dictionary or JSON file. All parameters are initialized
during its creation, while retaining values specified in the seed
objects or dictionary. The resulting object then maps to an exhaustive
input file required by RESCU+. It contains additional methods which may
provide valuable information about the system (e.g. number of atoms,
converting units). The structure is easily deduced from the API
documentation.
A typical input file (here GaAs-FCC) looks as follows:
from nanotools import Atoms, Cell, Kpoint, System, TotalEnergy
a = 2.818
cell = Cell(avec=[[0.,a,a],[a,0.,a],[a,a,0.]], resolution=0.12)
fxyz = [[0.00,0.00,0.00],[0.25,0.25,0.25]]
atoms = Atoms(fractional_positions=fxyz, formula="GaAs")
sys = System(cell=cell, atoms=atoms)
calc = TotalEnergy(sys)
calc.solve()
print(calc.energy.etot)
Atomic configurations
Atomic configurations consist in sets of coordinates and species. The
coordinates are specified either by the positions or
fractional_positions attribute of Atoms objects. The atomic
species are specified in the attribute formula of an Atoms
object. Each atom has a position and a corresponding label or symbol,
and each label corresponds to a species. In supercells, one can use the
following short hand instead of repeating symbols.
formula="GaGaGaGaGaGaGaGaGaGaGaGaGaGaGaGaAsAsAsAsAsAsAsAsAsAsAsAsAsAsAsAs"
is equivalent to
formula="Ga(16)As(16)"
For larger systems, one can simply specify the configuration in an
xyz-file and pass it to either the positions or
fractional_positions attribute. In this case formula need not be
set.
Pseudopotentials
nanotools can read pseudopotential (PS) files and numerical atomic orbital (NAO)
basis files generated by our atomic code Nanobase, i.e. the same PS/NAO files
as those with NanoDCAL and
RESCU distributions.
To install PS, untar the PS archive provided to you with the source code
in a known folder and set the environment variable RESCUPLUS_PSEUDO to that folder. Note that you may need to
change this depending on your applications: when using LDA you may set
RESCUPLUS_PSEUDO=path/to/lda/pseudos; when using PBE you may set
RESCUPLUS_PSEUDO=path/to/pbe/pseudos. Or simply copy the PS files
you will use to $PWD and set RESCUPLUS_PSEUDO=$PWD.
The DFT package RESCU+ and the NEGF-DFT package NanoDCAL+ are pseudopotential (PS) codes. They support the Troullier-Martin (TM) PS and the Optimized Norm-Conserving Vanderbilt (ONCV) PS.
Both RESCU+ and NanoDCAL+ use numerical atomic orbital (NAO) functions to form linear combination of atomic orbital basis (LCAO). These NAOs have been carefully tested using two methods. The first is the standard Δ-gauge which quantifies the difference of the equation of state (EOS) calculated by RESCU+ using LCAO basis and two benchmarks. One set of benchmark results was from the full potential WIEN2k package; while the second set of benchmark results was from the plane waves (PW) method of RESCU. Typically, Δ-values below 2 meV/atom suggests the LCAO basis to be excellent while somewhat higher values are also quite acceptable for many applications. The second test of the NAO is via a β-gauge that quantifies the difference of electronic band structures obtained by RESCU+ using LCAO basis and benchmark results, where the benchmarks were obtained from RESCU using the PW basis. The β-values have the same order of magnitude and unit as Δ, and a small β implies small deviation of band structures between LCAO and PW basis. In NEGF-DFT quantum transport simulations using NanoDCAL+ and/or many other applications, accurate band structures are critical.
In release 2023B, for ONCV, Triple Zeta Polarization (TZP), Double Zeta Polarization (DZP) and Single Zeta Polarization (SZP) NAO basis functions are provided. TZP is the largest basis followed by DZP and SZP. The Δ-values for TZP, DZP and SZP basis with respect to the WIEN2k benchmark using GGA-PBE, are shown in the following three periodic tables:
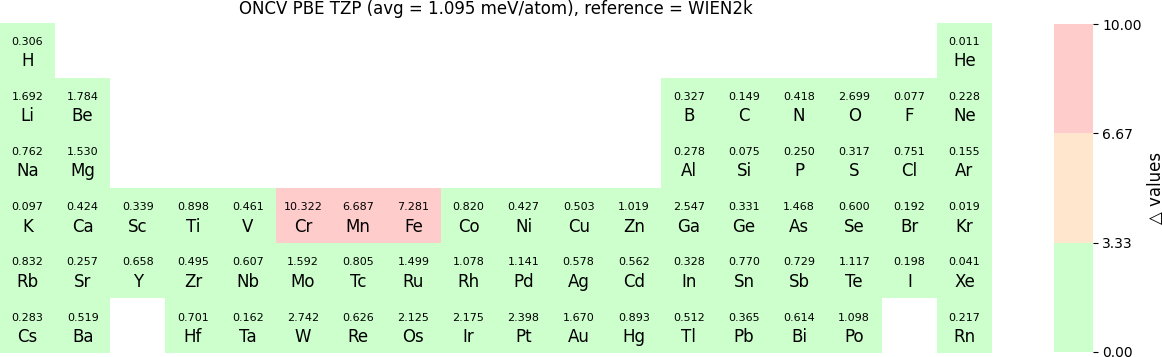
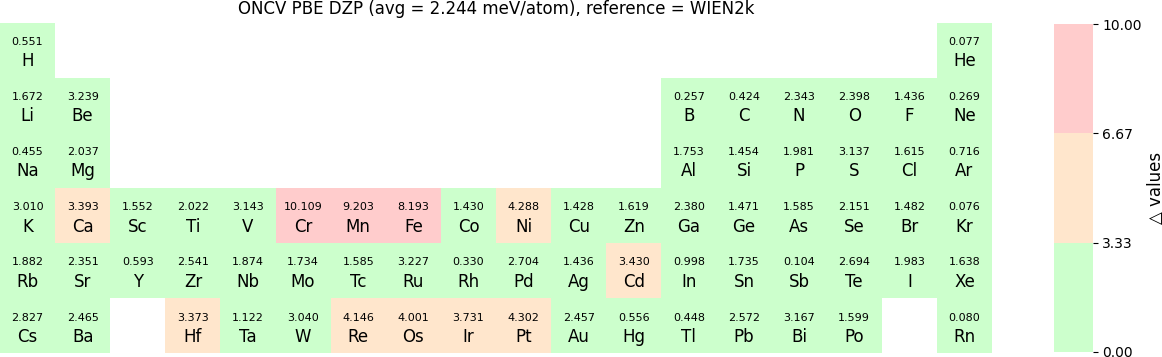
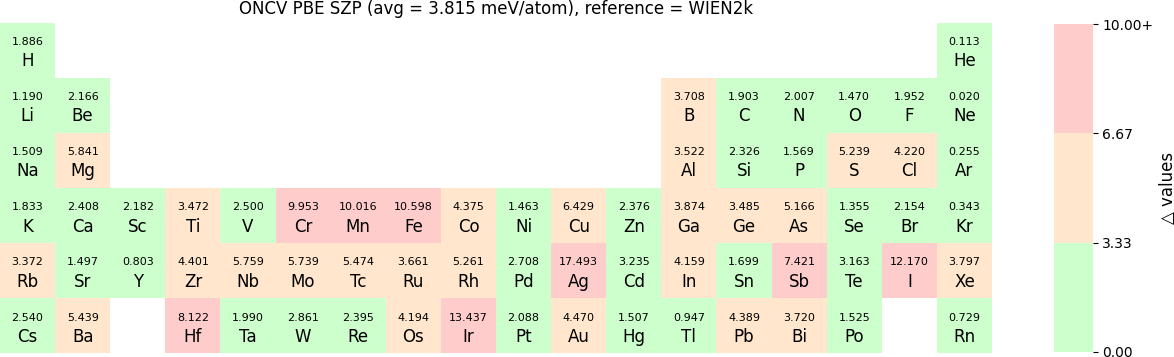
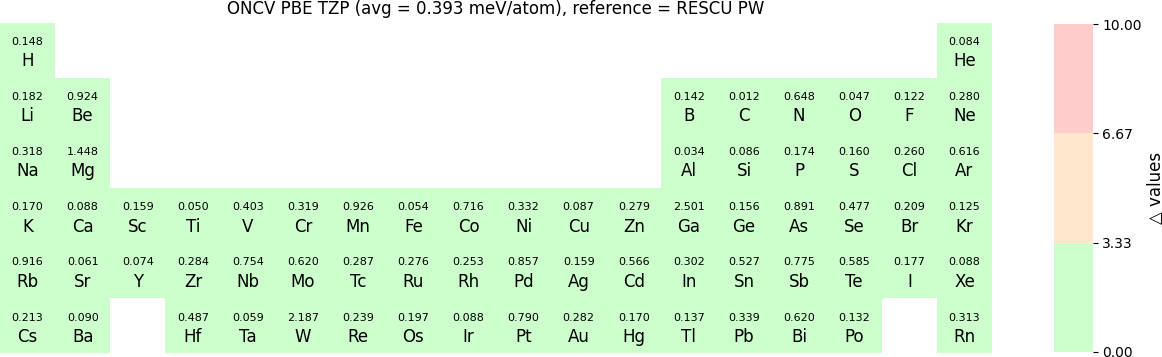
The Δ-values for the above NAO functions benchmarked with RESCU using PW basis are shown in the following three periodic tables:

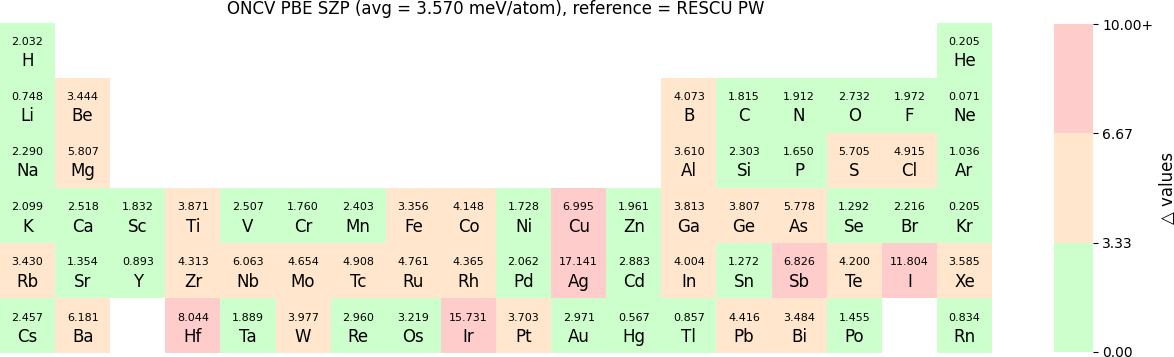
The β-values for the TZP, DZP and SZP NAO functions benchmarked with RESCU using PW basis, are shown in the following three periodic tables.
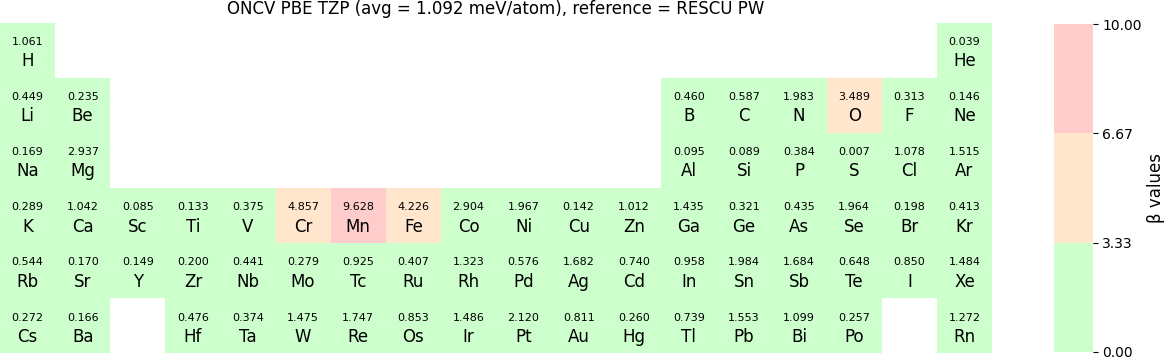
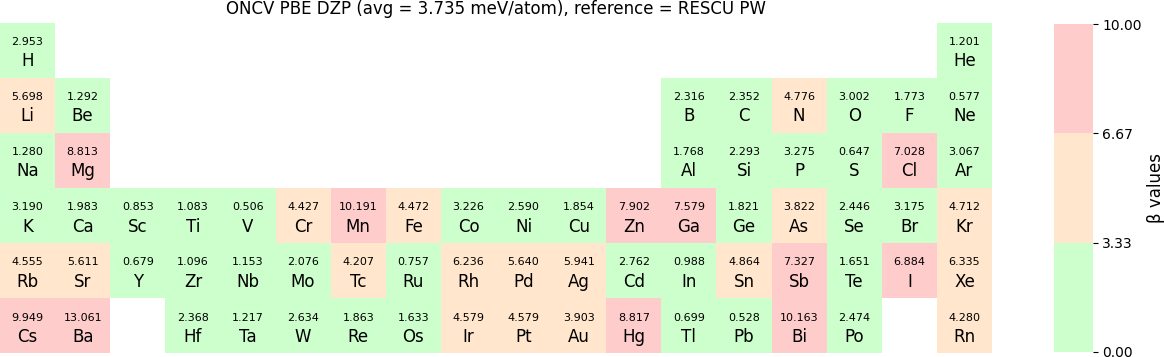
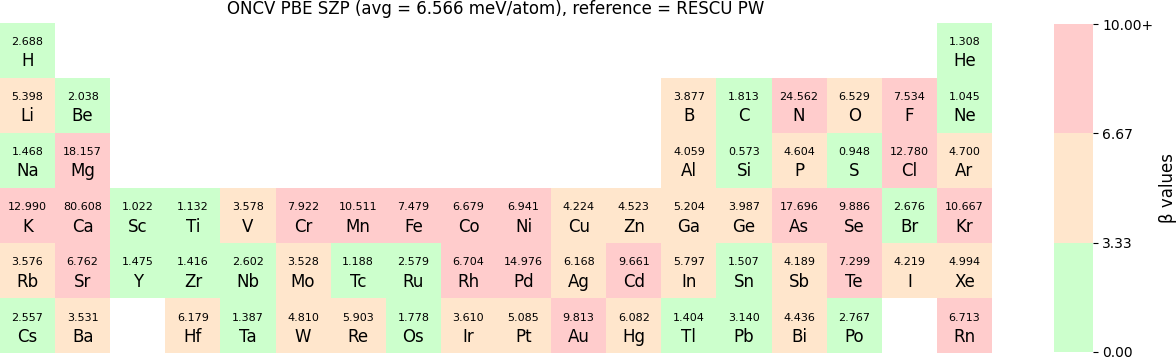
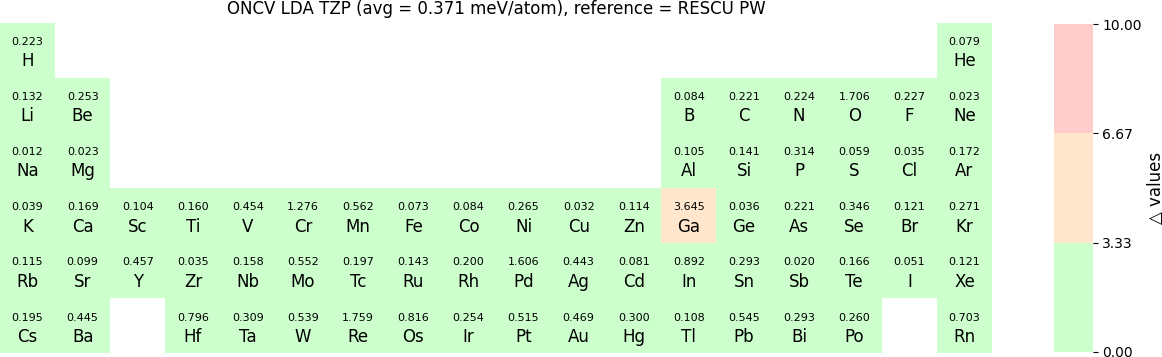
In release 2023B, TZP basis functions generated by the LDA exchange-correlation functional are also provided. Their Δ values benchmarked with WIEN2k and with RESCU using PW basis, are shown in the following two periodic tables:

\(\Delta\) values for TZP basis sets with respect to the PW method (LDA):
The β values for TZP basis sets with LDA benchmarked by RESCU using PW basis are shown in the following periodic table:

The following table provides a summary of the two metrics Δ and β in units of meV/atom for four NAO databases using ONCV PS. These databases were optimized using different exchange-correlation functionals and come in various sizes, including TZP, DZP and SZP. Δ (LCAO-WIEN2k) represents Δ values of the LCAO basis sets benchmarked with WIEN2k, Δ (LCAO-PW) represents Δ values benchmarked with RESCU using PW basis, and β (LCAO-PW) represents the β values between LCAO and PW basis. Mean values, averaged over 70 elements in the periodic table, of the two metrics and the corresponding standard deviations (SD) are provided for statistical purposes.
metric |
Δ (LCAO-WIEN2k) |
Δ (LCAO-PW) |
β (LCAO-PW) |
|||
|---|---|---|---|---|---|---|
statistics |
mean |
SD |
mean |
SD |
mean |
SD |
ONCV PBE TZP |
1.0947 |
1.6716 |
0.3933 |
0.4451 |
1.0924 |
1.4246 |
ONCV PBE DZP |
2.2439 |
1.8502 |
1.8317 |
1.0409 |
3.7351 |
2.8003 |
ONCV PBE SZP |
3.8149 |
3.2230 |
3.5696 |
3.0109 |
6.5664 |
10.026 |
ONCV LDA TZP |
1.1513 |
1.3435 |
0.3706 |
0.5464 |
1.1560 |
1.6726 |
While it is the responsibility of users to select the most proper LCAO basis for their applications, here are some general suggestions:
Experimenting the size of the basis sets (TZP, DZP or SZP) according to your system and goal. Larger basis sets may give more accurate results at the expense of computational time. Smaller basis sets can also give quite reasonable results for many applications. DZP basis sets are suggested for a balance between accuracy and computational cost.
Always test convergence before performing lengthy production calculations, by gradually increasing the real space grid, which corresponds to the mesh cut-off energy.
For stress tensor calculations of magnetic materials, fine real space grids should be tested (for example, 0.06 Bohr grid spacing for Fe, Co, Ni etc.) to attain precision.
Arrays
Many quantities are conveniently represented as arrays. For example, lattice vectors are written as
avec = [[1.,2.,3.],[4.,5.,6.],[7.,8.,9.]]
If arrays with more than one dimension are written as vectors, they will be reshaped automatically. We strongly suggest to explicitly specify the multidimensional array as above. If for some reason you need to employ one-dimensional arrays, read the following. RESCU+ uses the so-called Fortran order, which means that a vector is reshaped filling the 1st dimension, then the 2nd, then the 3rd etc. This means that
avec = [1.,2.,3.,4.,5.,6.,7.,8.,9.]
is reshaped to
avec = [[1.,4.,7.],[2.,5.,8.],[3.,6.,9.]]
which corresponds to a matrix
By contrast, there exist the so-called C ordering where multidimensional arrays are filled backward, i.e. starting by the last dimension.
This means that
avec = [1.,2.,3.,4.,5.,6.,7.,8.,9.]
is reshaped to
avec = [[1.,2.,3.],[4.,5.,6.],[7.,8.,9.]]
which corresponds to a matrix
Units
RESCU+ uses the Python library Pint to keep track of units. What you need to know to get started is that
Quantities with units in RESCU+ are Quantity instances.
You can get the magnitude of a Quantity object a with a.m.
You can get the unit of a Quantity object a with a.u.
You can import the unit registry from nanotools.utils, would you like to create Quantity objects of your own.
You may refer to the Pint documentation if you would like to know more about the library’s functionalities.
RESCU+ works with two unit systems:
extended SI: eV and \mathring{\text{A}}
atomic: hartree and bohr
The units of the
exhaustive input file are hartree and bohr by default, since these files
are intended for internal use of the Fortran code.
Python entities, like a seed dictionary or any data in a
TotalEnergy object, are in SI by default. Attributes which
are of the Quantity class are automatically converted on entry.
One can specify units in two ways:
use the unit registry from nanotools.utils.
pass a two-element tuple containing the magnitude and the unit as a string.
For example, in Listing 1, I can specify the units of avec as follows
from nanotools.utils import Quantity, ureg
a = 2.818
cell = Cell(avec=[[0.,a,a],[a,0.,a],[a,a,0.]] * ureg.angstrom, resolution=(0.2, "bohr"))
# [...]
print(calc.energy.etot.to("picojoules"))
First, avec is converted to a Quantity with units of angstrom multiplying by ureg.angstrom.
Then, resolution is specified in bohr, passing a tuple consisting of a float and the string bohr.
Return switches
To save time, a user may set return switches to perform or skip
certain calculations. For example the atomic forces, obtained upon
completing a ground state calculation, are not always necessary. It is
thus possible to set ["energy"]["forces_return"], which corresponds
to ["energy"]["forces"], to false and save time by skipping the
force calculation. Similarly, several output parameters have a
corresponding _return parameter.
Checking convergence
You can verify the convergence of your DFT calculation at the end of the process to ensure accuracy. Simply print the convergence status as shown in this example:
# Example code to check DFT calculation convergence
from ase.build import bulk
from nanotools import System, TotalEnergy, Relax
a = 5.43 # lattice constant in ang
atoms = bulk("Si", "diamond", a=a, cubic=True)
sys = System.from_ase_atoms(atoms)
sys.cell.set_resolution(0.2)
sys.kpoint.set_grid([5,5,5])
ecalc = TotalEnergy(sys)
ecalc.solver.set_mpi_command("mpiexec -n 2")
ecalc.solve()
print(ecalc.solver.mix.converged)
Output files
Upon the completion of a calculation, the software will automatically generate two output files in the working directory. Take total energy calculation as an example, the two output files are nano_scf_out.json and nano_scf_out.h5. These files contain important information about the simulation and results. They can be used for post-processing and analysis. The details of each file are as follows:
nano_scf_out.json
This file stores structured metadata and results of the calculation in a JSON format, providing easy access to important quantities. The following objects can be found inside the JSON file:
“classname”
This key defines the type of calculation performed. Possible values are “TotalEnergy”, “BandStructure”, “DensityOfStates”, etc.
“energy”
This object stores the energy-related results: the computed various types of energy of the system (refer to nanotools.energy module), eigenvalues of the electronic states, atomic forces and stress tensor if the corresponding return keyword is set to True.
“solver”
Information related to the solver and computation details can be found here, including: the basis type used in the calculation, information about the executable binary and licensing, details of the eigenvalue solver used, mixing parameters, information about the MPI processes and blocking, and options for restarting the calculation.
“system”
This object contains the system’s simulation parameters: the atomic species and their positions, the lattice vectors, the boundary conditions applied to the system, the grid number of k-points used for Brillouin zone sampling, the resolution of the real space grid, and the XC functional used in the DFT calculation.
nano_scf_out.h5
This file stores the result data in HDF5 format. It contains more detailed or larger datasets that might not be efficiently stored in a JSON file. One can use the h5py library to read the data from the HDF5 file.
import h5py
with h5py.File("nano_scf_out.h5", "r") as f:
print(f.keys())
<KeysViewHDF5 ['density', 'potential']>
By default, the file contains the following datasets:
“density”
<KeysViewHDF5 ['ispin', 'neutralatom', 'partialcore', 'total']>
As shown above, the “density” dataset contains four sub-datasets: “ispin”, “neutralatom”, “partialcore”, and “total”. The “ispin” dataset stores an integer indicating the spin type of the system. The “neutralatom” dataset stores the neutral atom density. The “partialcore” dataset stores the partial core density. The “total” dataset stores the total electron density. Since electron densities (total, neutral, and partial core) are evaluated on the same grid of the computational domain, they have the same shape of (nu, nv, nw), where nu, nv, and nw are the number of grid points along dimension #1, #2 and #3, respectively.
“potential”
<KeysViewHDF5 ['deltahartree', 'effective', 'external', 'ispin', 'neutralatom', 'xc']>
The “potential” dataset contains five sub-datasets: “deltahartree”, “effective”, “external”, “ispin”, “neutralatom”, and “xc”. The “deltahartree” dataset stores the Hartree potential. The “effective” dataset stores the effective potential. The “external” dataset stores the external potential. The “ispin” dataset stores an integer indicating the spin type of the system. The “neutralatom” dataset stores the neutral atom potential. The “xc” dataset stores the exchange-correlation potential.