Scattering states tutorial
Requirements
Software components
nanotools
NanoDCAL+
Pseudopotentials
C_LDA_TM_DZP.mat
Copy the mat file from the pseudo database to the current working
directory and export NANODCALPLUS_PSEUDO=$PWD.
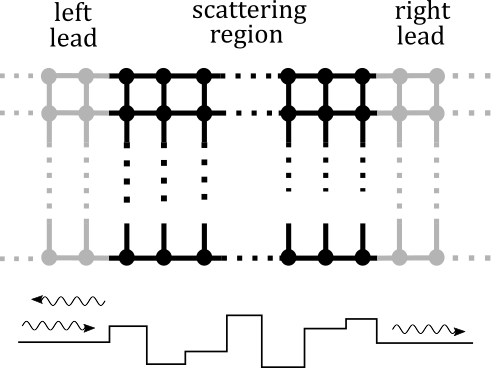
Fig. 1 A two-probe transport system
Briefing
Scattering states are eigenstates of a quantum transport system. Given an incident wave from one lead of the device, we wish to compute its scattering rates into other outgoing waves at the same energy level, together with the scattered wave amplitude in the center of the device. To be concrete, the program solves the following linear system of equations [1]
where \Sigma represents lead self-energies, S is a source term arising from incident waves, and the solution to this equation, i.e. \phi_1 \cdots \phi_n, are coefficients of the scattered wavefunction. The reflection and the transmission coefficients are then calculated from the following equations [2]
since the reflected and the transmitted waves must be linear combinations of outgoing waves in the leads, whose coefficients are put as matrix-columns in \mathbf{\Phi}_L^- and \mathbf{\Phi}_R^+.
This tutorial demonstrates the computational workflow by analyzing a carbon chain system with an anti-parallel spin configuration in the leads.
Scripts
left.py
from nanotools import Atoms, Cell, System, TotalEnergy
with open("left.xyz", "w") as f:
f.write(
"""3
s x y z sz
C 6.00000000 6.00000000 7.25000000 0.5
C 6.00000000 6.00000000 4.35000000 0.5
C 6.0 6.00000000 1.45000000 0.5
""")
npc = 4
cell = Cell(avec=[12, 0.0, 0.0,
0.0, 12, 0.0,
0.0 , 0.0 , 8.7],
grid =[92, 92, 68])
atoms= Atoms(positions="left.xyz")
sys = System(cell=cell, atoms=atoms)
sys.hamiltonian.ispin = 2
sys.atoms.set_initial_magnetic_moments("left.xyz")
sys.kpoint.set_grid([1,1,100])
sys.pop.type = "fd"
sys.pop.set_sigma(0.01)
etot = TotalEnergy(sys)
etot.solver.mpidist.kptprc = npc
etot.solver.cmd.mpi = f"mpiexec -n {npc}"
etot.solver.restart.densityPath = "left_out.h5"
etot.solve(input="left", output="left_out")
right.py
from nanotools import Atoms, Cell, System, TotalEnergy
with open("right.xyz", "w") as f:
f.write(
"""3
s x y z sz
C 6.00000000 6.00000000 7.25000000 -0.5
C 6.00000000 6.00000000 4.35000000 -0.5
C 6.0 6.00000000 1.45000000 -0.5
""")
npc = 4
cell = Cell(avec=[12, 0.0, 0.0,
0.0, 12, 0.0,
0.0 , 0.0 , 8.7],
grid =[92, 92, 68])
atoms= Atoms(positions="right.xyz")
sys = System(cell=cell, atoms=atoms)
sys.hamiltonian.ispin = 2
sys.atoms.set_initial_magnetic_moments("right.xyz")
sys.kpoint.set_grid([1,1,100])
sys.pop.type = "fd"
sys.pop.set_sigma(0.01)
etot = TotalEnergy(sys)
etot.solver.mpidist.kptprc = npc
etot.solver.cmd.mpi = f"mpiexec -n {npc}"
etot.solver.restart.densityPath = "right_out.h5"
etot.solve(input="right", output="right_out")
center.py
from nanotools import Atoms, Cell, System, TotalEnergy, Transmission, TwoProbe
import numpy as np
with open("center.xyz", "w") as f:
f.write(
"""12
s x y z sz
C 6.00000000 6.00000000 33.35000000 1.5
C 6.00000000 6.00000000 24.65000000 1.5
C 6.00000000 6.00000000 15.95000000 1.5
C 6.00000000 6.00000000 7.25000000 1.5
C 6.00000000 6.00000000 30.45000000 1.5
C 6.00000000 6.00000000 21.75000000 1.5
C 6.00000000 6.00000000 13.05000000 1.5
C 6.00000000 6.00000000 4.35000000 1.5
C 6.00000000 6.00000000 27.55000000 1.5
C 6.00000000 6.00000000 18.850000 1.5
C 6.00000000 6.00000000 10.15000000 1.5
C 6.00000000 6.00000000 1.45000000 1.5
""")
left = TotalEnergy.read(filename="left_out.json")
right= TotalEnergy.read(filename="right_out.json")
cell = Cell(avec=[12, 0.0, 0.0,
0.0, 12, 0.0,
0.0 , 0.0 , 34.8],
grid=[92, 92, 272])
atoms= Atoms(positions="center.xyz")
sys = System(cell=cell, atoms=atoms)
sys.hamiltonian.ispin = 2
sys.atoms.set_initial_magnetic_moments("center.xyz")
sys.kpoint.set_grid([1,1,1])
sys.pop.type = "fd"
sys.pop.sigma= 0.01
sys.pop.nPole= 20
center = TotalEnergy(sys)
npc = 8
dev = TwoProbe(left=left, center=center, right=right, transport_axis=2)
dev.center.solver.cmd.mpi = f"mpiexec -n {npc}"
dev.center.solver.mix.alpha = 0.1
dev.center.solver.restart.densityPath = "center_out.h5"
dev.center.solver.mix.precond = "kerker"
# dev.center.solver.mpidist.zptprc = npc
dev.center.solver.mix.maxit = 100
dev.solve()
transm.py
import numpy as np
from nanotools import TwoProbe, Transmission
dev = TwoProbe.read(filename="nano_2prb_out.json")
trs = Transmission.from_twoprobe(twoprb=dev)
trs.solve(energies = np.linspace(0, 1.6, num=200))
trs.plot(filename="transm.png")
scatt.py
from nanotools import TwoProbe
from nanotools.scattering import ScatteringStates
dev = TwoProbe.read(filename="nano_2prb_out.json")
dev.center.system.kpoint.set_fractional_coordinates([[0,0,0]])
sct = ScatteringStates.from_twoprobe(dev)
sct.solve(energy=1.2) # compute scattering states at 1.2eV
analy.py
from nanotools import ScatteringStates
calc = ScatteringStates.read("nano_scatt_in.json")
incoming= calc.get_bloch(lead="left", direction="in", k_long_index=0)
outgoing= calc.get_bloch(lead="right", direction="out", k_long_index=0)
t = abs(calc.get_scatter_rate(incoming, outgoing))**2 # t=0.302
calc.plot_isosurfaces(**incoming, vals=[1.])
wave = calc.get_scatt_wave(**incoming)
Explanations
The physical system consists of a chain of carbon atoms with equal
distances. We divide the chain into three regions, i.e. left, center,
and right. We start with the ground-state calculation for left lead by
running left.py, where the spin polarization is set upward. Next we
run right.py for the right lead, with downward spin. After getting
the ground states for leads, we proceed with the two probe calculation
by running center.py. To gain a general understanding of the
transport properties of our device, we compute its transmission using
transm.py. The result is presented in the following figure.
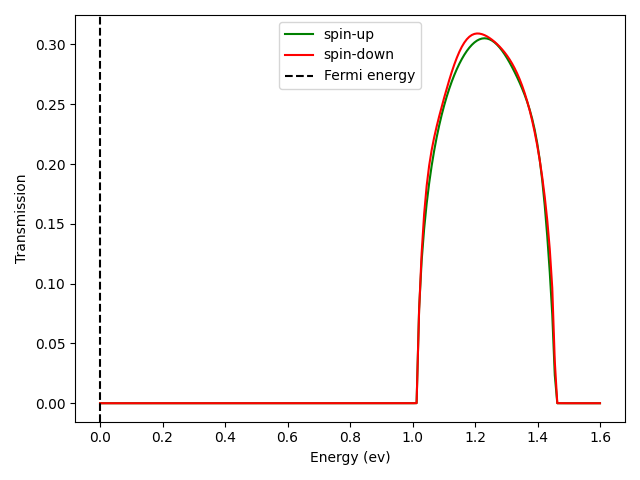
Fig. 2 Transmission profile of carbon chain
We then compute the scattering states (scatt.py) at the energy level
1.2eV. The result is saved in nano_scatt_out.h5 file, which contains
information about incoming and outgoing waves in both leads, their
scattering rates, and corresponding wave-functions in the central
scattering region of the device. To ease access to the data, we have
implemented a few methods in the ScatteringStates class. The following
script extracts information on a pair of incoming/outgoing Bloch-waves
hosted by the left and the right leads respectively.
from nanotools import ScatteringStates
calc = ScatteringStates.read("nano_scatt_in.json")
incoming = calc.get_bloch(lead="left", direction="in", k_long_index=0)
outgoing = calc.get_bloch(lead="right", direction="out", k_long_index=0)
The get_bloch method returns a dictionary for incoming:
{'lead': 'left',
'direction': 'in',
'spin': 1,
'spin_direction': 'up',
'k_trans_frac': array([0, 0, 0]),
'k_trans_index': 0,
'k_long_index': 0,
'k_long_frac': -0.25836461407263456,
'energy': 1.2,
'energy_index': 0
}
Note that there can be more than one propagating wave at a given energy;
if we don’t specify which wave through parameter k_long_index, all
the longitudinal wave numbers will be listed in k_long_frac.
One important piece of information that we wish to know is the
scattering rate from one incoming wave to another outgoing wave. This
piece of information is stored in the scatt_mat_out_in field in the
nano_scatt_out.h5 output file, and can be read out with the
get_scatter_rate method:
t = calc.get_scatter_rate(incoming, outgoing) # t = -0.141 - 0.532j
abs(t)**2 # |t|^2 = 0.302
We notice that |t|^2 is just the transmission value at E=1.2eV
(see the transmission plot above), as there happens to be only one pair
of incoming and outgoing states at the given energy and spin.
The scattered wave that originates from incoming can be obtained, as
a 3d array with values defined on the real space grid, via
wave = calc.get_scatt_wave(**incoming)
The wave-function can be visualized with
calc.plot_isosurfaces(**incoming, vals=[1.])
where we plot its isosurface at absolute value 1.0. Multiple isosurfaces
can be plotted when vals is a list of multiple values.
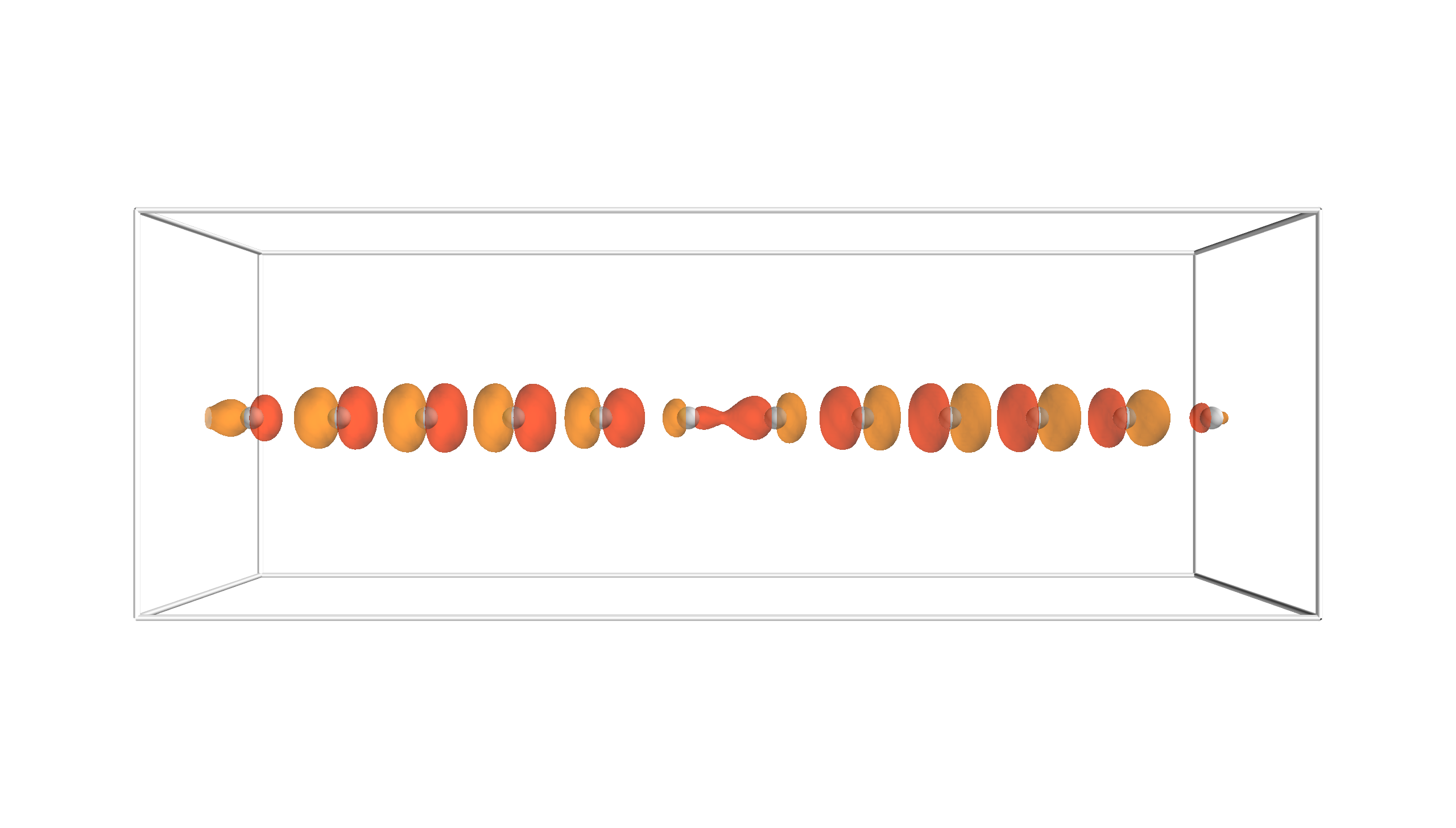
Fig. 3 Scattered wave carbon chain