Transmission tutorial
Requirements
Software components
nanotools
NanoDCAL+
Pseudopotentials
C_AtomicData.mat
Copy the mat file from the pseudo database to the current working directory and export NANODCALPLUS_PSEUDO=$PWD.
Briefing
This tutorial presents a minimal example of transmission calculation for a two-probe transport system. The system to be addressed is a crystalline carbon nano-tube (CNT).
Setup
python script
from nanotools import Atoms, Cell, Species, System, TotalEnergy, Transmission, TwoProbe
import numpy as np
cell = Cell(avec=[[20.0, 0.0, 0.0], [0.0, 20.0, 0.0], [0.0 , 0.0 , 2.4595]], resolution=0.2)
atoms= Atoms(positions="cnt.xyz")
sys = System(cell=cell, atoms=atoms)
sys.kpoint.set_grid([1,1,50])
center = TotalEnergy(sys)
left = center
right= center
dev = TwoProbe(left=left, center=center, right=right, transport_axis=2)
dev.center.solver.set_mpi_command("mpiexec -n 4")
# dev.center.solver.mpidist.zptprc=4
dev.solve()
cal = Transmission.from_twoprobe(twoprb=dev)
cal.solve(energies = np.linspace(-5, 5, num=101))
cal.plot(filename="cnt_transm.png", show=True)
structure file cnt.xyz, in Cartesian coordinates
12
CNT
C 8.08866505 10.69566903 0.61487804
C 8.44186547 11.30743011 1.84462196
C 9.64679958 12.00309914 1.84462196
C 10.35320042 12.00309914 0.61487804
C 11.55813453 11.30743011 0.61487804
C 11.91133495 10.69566903 1.84462196
C 11.91133495 9.30433097 1.84462196
C 11.55813453 8.69256989 0.61487804
C 10.35320042 7.99690086 0.61487804
C 9.64679958 7.99690086 1.84462196
C 8.44186547 8.69256989 1.84462196
C 8.08866505 9.30433097 0.61487804
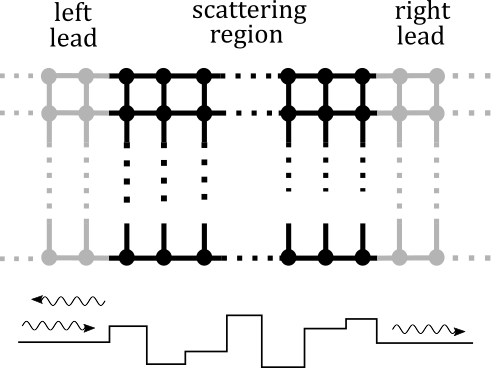
Explanations
As sketched in the above diagram, a two-probe transport system consists of three sub-systems, corresponding to the two electrical leads and a quantum scattering structure in between. The leads are regarded as in thermal equilibrium and therefore can be resolved with the basic DFT calculator under periodic boundary conditions. The remaining task is solving the quantum scattering problem with lead electronic structures taken as boundary conditions. We shall first perform a self-consistent calculation for our CNT structure, and then compute the very important quantity, transmission, which indicates the maximum possible electric current at a given energy level.
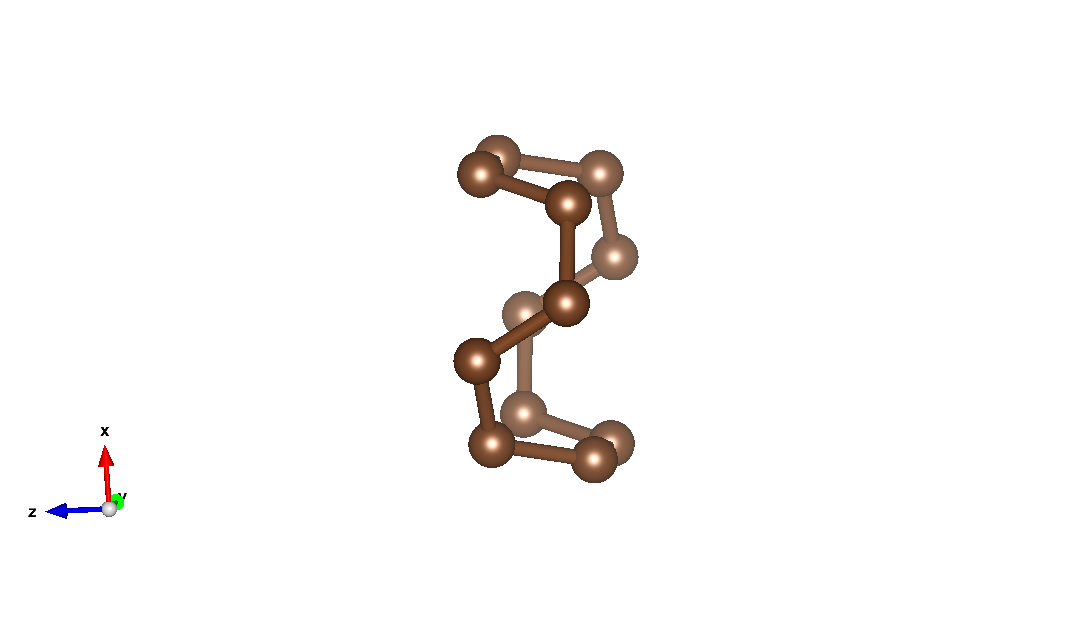
System definition
The following code section describes a unit cell of an armchair carbon nano-tube (see figure above)
cell = Cell(avec=[[20.0, 0.0, 0.0], [0.0, 20.0, 0.0], [0.0 , 0.0 , 2.4595]], resolution=0.2)
atoms= Atoms(positions="cnt.xyz")
sys = System(cell=cell, atoms=atoms)
A grid of reciprocal (k) points is also needed for the periodic calculation of lead structures. This is specified by
sys.kpoint.set_grid([1,1,50])
We set only one k-point in the transversal plane since there is vacuum buffer all around the nanotube. Along the transport direction, on the other hand, a sufficient number of k-points should be used since lead structures are periodic. In general, only one k-grid needs be specified for the whole two-probe calculation: k-points along the transport dimension are automatically removed when it comes to the NEGF calculation for the central region.
The sys object is then used to initialize a TotalEnergy calculator (see tutorial_etot.md)
center = TotalEnergy(sys)
The TwoProbe calculator, which manages the work-flow of self-consistent calculations,
can be initialized as
left = center
right= center
dev = TwoProbe(left=left, center=center, right=right, transport_axis=2)
where left, center, and right are TotalEnergy calculators for the respective sub-systems. In this
tutorial, the three sub-systems are structually identical. Therefore, we simply assign the same calculator twice
Since it happens quite often that a few or all of the sub-systems in a two-probe system share the same structure, nanotools provides a simplified interface for this case
dev = TwoProbe(center=center, transport_axis=2)
Now left and right are assumed identical to center.
When only right is omitted, eg.
dev = TwoProbe(left=left, center=center, transport_axis=2)
right is assumed identical to left.
Nevertheless, note that one must explicitly specify the transport_axis parameter
(along which dimension transport occurs) in defining a TwoProbe calculator.
In the current example, transport_axis=2 corresponds to the z-dimension
(remember that python uses 0-based indexing).
Self-consistent calculation
We then launch the self-consistent calculation by typing in
dev.center.solver.set_mpi_command("mpiexec -n 4")
dev.center.solver.mpidist.zptprc=4
dev.solve()
The first two lines set mpi-related parameters to accelerate the calculation.
Here we assign 4 processors. The zptprc parameter indicates the number of
energy points at which Green functions are computed in parallel. This parameter
is better to be maximized for small simulation tasks.
Upon executing dev.solve(), a few json files will appear in the directory; they are left.json, right.json, and nano_2prb_in.json.
In many cases the leads may be structurally identical; if so, right.json will be omitted.
Self-consistent calculations are first carried out for leads, using the rescuplus executable, with left.json or right.json
as its input files. The outputs are written in left_out.json, left_out.h5, right_out.json and right_out.h5.
After the lead calculations are finished, nanodcalplus will be executed with nano_2prb_in.json as its input file.
nanodcalplus performs the self-consistent calculation for the quantum scattering problem, and the results
will be saved in nano_2prb_out.json and center_out.h5. Note that center_out.h5 contains charge densities
and potentials only of the central scattering region.
All the workflow described here is managed by dev.solve().
One may restart a two-probe calculation based on an existing charge-density file, e.g.
...
dev.center.solver.restart.densityPath = "center_out.h5"
dev.solve()
Transmission
The self-consistent calculation must precedes the transmission calculation, since the latter requires
existing ground-state density data, i.e. _out.h5 files.
The Transmission calculator is dedicated to the transmission computation. In our case,
we can simply create such a calculator from the existing TwoProbe object and use its
solve method to launch the calculation.
cal = Transmission.from_twoprobe(twoprb=dev)
cal.solve(energies = np.linspace(-5, 5, num=101))
Finally, we visualize the transmission profile by typing in
cal.plot(filename="cnt_transm.png", show=True)
You should see something similar to
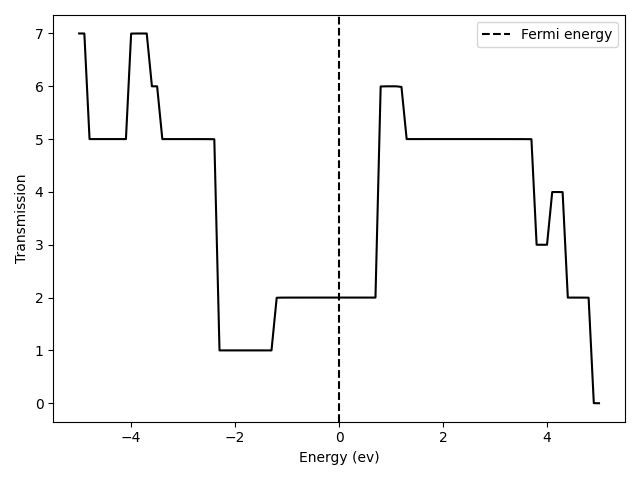
Since we are simulating a perfectly intrinsic carbon nano-tube, its transmission profile shows a step-wise shape. The integer transmission value at each step simply equals the band degeneracy.