Spintronics tutorial
Requirements
Software components
nanotools
NanoDCAL+
References
J.Maassen, W. Ji and H. Guo, Nano. Lett., 11, 151 (2011)
Pseudopotentials
C_LDA_TM_DZP.mat
Ni_LDA_TM_DZP.mat
Copy the mat files from the pseudo database to the current working directory and export NANODCALPLUS_PSEUDO=$PWD.
Structure files
c_ni_left.xyz
c_ni_right.xyz
c_ni_center.xyz
Briefing
In this tutorial, we simulate the transport property of a graphene - spintronic device consisting of a single graphene sheet embedded in spin-polarized nickel. The model is sketched in the figure below. Transport occurs along the y-axis, and the structure is periodic along x,z-axes.
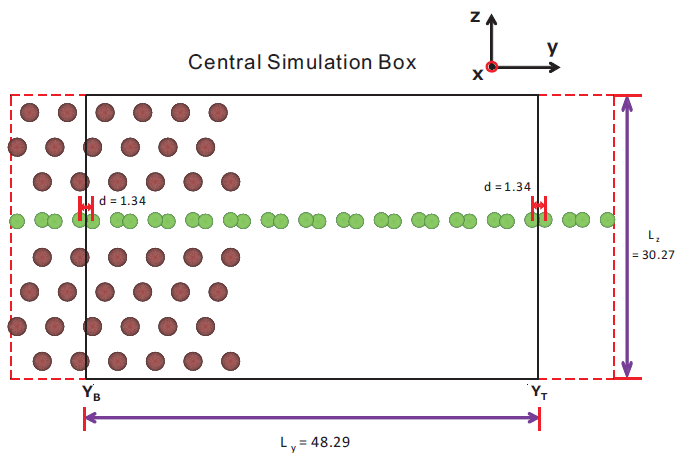
Script
The full python script for our calculation is as follows:
from nanotools import Atoms, Cell, System, TotalEnergy, Transmission, TwoProbe
npc = 40 # number of processors to be used
# set up TotalEnergy calculator for left lead
cell = Cell(
avec=(
[
[4.64682193271677, 0.0, 0.0],
[0.0, 8.04853168139326, 0.0],
[0.0, 0.0, 30.27076472739540],
],
"bohr",
),
resolution=(0.2, "bohr"),
)
atoms = Atoms(positions=("c_ni_left.xyz", "bohr"))
sys = System(cell=cell, atoms=atoms)
sys.hamiltonian.ispin = 2
sys.atoms.set_initial_magnetic_moments("c_ni_left.xyz")
sys.kpoint.set_grid([10, 40, 1])
sys.pop.type = "fd"
left = TotalEnergy(sys)
left.solver.mix.alpha = 0.02
left.solver.mix.precond = "kerker"
left.solver.mix.maxit = 150
left.solver.mix.tol = 1.0e-6
left.solver.restart.densityPath = "left_out.h5"
left.solver.set_mpi_command(f"mpiexec -n {npc}")
left.solver.mpidist.kptprc = npc
# set up TotalEnergy calculator for right lead
cell = Cell(
avec=(
[
[4.64682193271677, 0.0, 0.0],
[0.0, 8.04853168139326, 0.0],
[0.0, 0.0, 30.27076472739540],
],
"bohr",
),
resolution=(0.2, "bohr"),
)
atoms = Atoms(positions=("c_ni_right.xyz", "bohr"))
sys = System(cell=cell, atoms=atoms)
sys.hamiltonian.ispin = 2
sys.pop.type = "fd"
sys.atoms.set_initial_magnetic_moments("c_ni_right.xyz")
sys.kpoint.set_grid([10, 40, 1])
right = TotalEnergy(sys)
right.solver.mpidist.kptprc = npc
right.solver.set_mpi_command(f"mpiexec -n {npc}")
# set up TotalEnergy calculator for central region
cell = Cell(
avec=(
[
[4.64682193271677, 0.0, 0.0],
[0.0, 48.29119008835956, 0.0],
[0.0, 0.0, 30.27076472739540],
],
"bohr",
),
resolution=(0.2, "bohr"),
)
atoms = Atoms(positions=("c_ni_center.xyz", "bohr"))
sys = System(cell=cell, atoms=atoms)
sys.hamiltonian.ispin = 2
sys.atoms.set_initial_magnetic_moments("c_ni_center.xyz")
sys.kpoint.set_grid([10, 1, 1])
sys.pop.type = "fd"
center = TotalEnergy(sys)
# solver parameters
center.solver.mix.alpha = 0.02
center.solver.mix.precond = "kerker"
center.solver.mix.maxit = 300
center.solver.mix.tol = 1.0e-6
center.solver.mpidist.kptprc = 10
center.solver.set_mpi_command(f"mpiexec -n {npc}")
center.solver.restart.densityPath = "center_out.h5"
# center.solver.cache_self_energy = False #default: True
# initialize TwoProbe calculator and perform self-consistent calculation
# transport occurs along y-axis (transport_axis=1)
dev = TwoProbe(left=left, center=center, right=right, transport_axis=1)
dev.solve()
# transmission calculation
# initialized transmission calculator from that for twoprobe
trs = Transmission.from_twoprobe(twoprb=dev)
# increase k-point mesh density
trs.center.system.kpoint.set_grid([120, 1, 20])
# set mpi parameters
trs.center.solver.mpidist.kptprc = npc
trs.center.solver.set_mpi_command(f"mpiexec -n {npc}")
# compute transmission at 0.1 eV
trs.solve(energies=0.1)
trs.plot(filename="spin_trsm.png")
structure file c_ni_left.xyz, in Cartesian coordinates
18
s x y z sz
Ni 1.16170546635838 0.67334134069663 5.56546536369770 1
Ni 1.16170546635838 0.68561034069663 24.71659336369770 1
Ni 3.48511646635838 2.00966934069663 9.25741636369770 1
Ni 3.48511646635838 2.01767134069663 28.41489436369770 1
Ni 1.16170546635838 3.35594834069663 12.95920936369770 1
Ni 1.16170546635838 3.35776134069663 1.85587036369770 1
Ni 1.16170546635838 3.36359234069663 21.01628036369770 1
Ni 3.48511646635838 4.69760734069663 5.56546536369770 1
Ni 3.48511646635838 4.70987734069663 24.71659336369770 1
Ni 1.16170546635838 6.03393534069663 9.25741636369770 1
Ni 1.16170546635838 6.04193834069663 28.41489436369770 1
Ni 3.48511646635838 7.38021634069663 12.95920936369770 1
Ni 3.48511646635838 7.38203034069663 1.85587036369770 1
Ni 3.48511646635838 7.38786034069663 21.01628036369770 1
C 1.16170546635838 0.67050034069663 16.79453236369770 1
C 1.16170546635838 3.35376234069663 16.95164236369770 1
C 3.48511646635838 4.69476834069663 16.79453236369770 1
C 3.48511646635838 7.37803134069663 16.95164236369770 1
structure file c_ni_right.xyz, in Cartesian coordinates
4
s x y z sz
C 1.16170546635838 3.35586034069663 16.96215117189000 0.0
C 1.16170546635838 0.67290534069663 16.96711717189000 0.0
C 3.48511646635838 7.38012834069663 16.96215117189000 0.0
C 3.48511646635838 4.69717334069663 16.96711717189000 0.0
structure file c_ni_center.xyz, in Cartesian coordinates
52
s x y z sz
Ni 1.16170546635838 0.67334134069663 5.56546536369770 0.5
Ni 1.16170546635838 0.68561034069663 24.71659336369770 0.5
Ni 3.48511646635838 2.00966934069663 9.25741636369770 0.5
Ni 3.48511646635838 2.01767134069663 28.41489436369770 0.5
Ni 1.16170546635838 3.35594834069663 12.95920936369770 0.5
Ni 1.16170546635838 3.35776134069663 1.85587036369770 0.5
Ni 1.16170546635838 3.36359234069663 21.01628036369770 0.5
Ni 3.48511646635838 4.69760734069663 5.56546536369770 0.5
Ni 3.48511646635838 4.70987734069663 24.71659336369770 0.5
Ni 1.16170546635838 6.03393534069663 9.25741636369770 0.5
Ni 1.16170546635838 6.04193834069663 28.41489436369770 0.5
Ni 3.48511646635838 7.38021634069663 12.95920936369770 0.5
Ni 3.48511646635838 7.38203034069663 1.85587036369770 0.5
Ni 3.48511646635838 7.38786034069663 21.01628036369770 0.5
Ni 1.16170546635838 8.72187302208989 5.56546536369770 0.5
Ni 1.16170546635838 8.73414202208989 24.71659336369770 0.5
Ni 3.48511646635838 10.05820102208989 9.25741636369770 0.5
Ni 3.48511646635838 10.06620302208989 28.41489436369770 0.5
Ni 1.16170546635838 11.40448002208989 12.95920936369770 0.5
Ni 1.16170546635838 11.40629302208989 1.85587036369770 0.5
Ni 1.16170546635838 11.41212402208989 21.01628036369770 0.5
Ni 3.48511646635838 12.74613902208989 5.56546536369770 0.5
Ni 3.48511646635838 12.75840902208989 24.71659336369770 0.5
Ni 1.16170546635838 14.08246702208989 9.25741636369770 0.5
Ni 1.16170546635838 14.09047002208989 28.41489436369770 0.5
Ni 3.48511646635838 15.42874802208989 12.95920936369770 0.5
Ni 3.48511646635838 15.43056202208989 1.85587036369770 0.5
Ni 3.48511646635838 15.43639202208989 21.01628036369770 0.5
C 1.16170546635838 0.67050034069663 16.79453236369770 0.05
C 1.16170546635838 3.35376234069663 16.95164236369770 0.05
C 3.48511646635838 4.69476834069663 16.79453236369770 0.05
C 3.48511646635838 7.37803134069663 16.95164236369770 0.05
C 1.16170546635838 8.71903202208989 16.79453236369770 0.05
C 1.16170546635838 11.40229402208989 16.95164236369770 0.05
C 3.48511646635838 12.74330002208989 16.79453236369770 0.05
C 3.48511646635838 15.42656302208989 16.95164236369770 0.05
C 1.16170546635838 16.76756370348315 16.79453236369770 0.00
C 1.16170546635838 19.45082570348315 16.95164236369770 0.00
C 3.48511646635838 20.79183170348315 16.79453236369770 0.00
C 3.48511646635838 23.47509470348315 16.95164236369770 0.00
C 1.16170546635838 24.81609538487641 16.79453236369770 0.00
C 1.16170546635838 27.49935738487641 16.95164236369770 0.00
C 3.48511646635838 28.84036338487641 16.79453236369770 0.00
C 3.48511646635838 31.52362638487641 16.95164236369770 0.00
C 1.16170546635838 32.86462706626967 16.79453236369770 0.00
C 1.16170546635838 35.54788906626968 16.95164236369770 0.00
C 3.48511646635838 36.88889506626967 16.79453236369770 0.00
C 3.48511646635838 39.57215806626967 16.95164236369770 0.00
C 1.16170546635838 40.91315874766293 16.79453236369770 0.00
C 1.16170546635838 43.59642074766293 16.95164236369770 0.00
C 3.48511646635838 44.93742674766293 16.79453236369770 0.00
C 3.48511646635838 47.62068974766294 16.95164236369770 0.00
Explanations
The script starts by defining TotalEnergy calculators for the three separate regions
of the transport structure. For each region we have one .xyz file to specify the atomistic
structure, where initial magnetic moments at individual atomic sites are also
specified in the sz column.
Collinear spin polarization is turned on by
sys.hamiltonian.ispin = 2
As for solver parameters we specify using the Kerker mixing algorithm, which is generally
preferable for metallic systems such as ours.
Note that in this tutorial we have enabled (by default) the program to cache lead self-energies so as to avoid computing them repetitively on the fly. However, under limited memory resource, one may want to disable this caching feature by
center.solver.cache_self_energy = False
In doing so, each iteration during the self-consistent calculation may take significantly longer time.
The self-consistent calculation typically takes many iterations to converge. If convergence
is not reached after mix.maxit iterations, run the following script to restart the calculation
from nanotools import TwoProbe
dev = TwoProbe.read(filename="nano_2prb_in.json")
dev.center.solver.mix.alpha = 0.05 # play with it!
dev.center.solver.restart.densityPath = "center_out.h5"
dev.solve()
Adjusting the mix.alpha parameter may also help.
The last part of our script is dedicated to transmission calculation. The transmission of our system is supposed to be a three dimensional function along the two periodic axes plus one energy dimension. For illustrative purpose we only compute the transmission at a single energy point. The resulting plot, saved as “spin_trsm.png” in file system, should look like the following
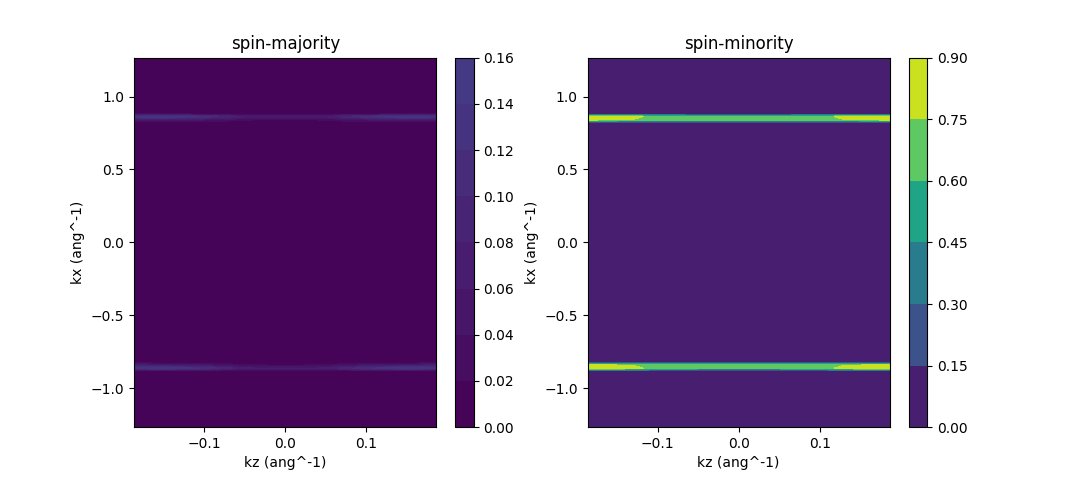
One may also use the following script to launch a transmission calculation, separate from the self-consistent calculation:
from nanotools import TwoProbe, Transmission
dev = TwoProbe.read(filename="nano_2prb_out.json")
trs = Transmission.from_twoprobe(twoprb=dev)
trs.center.system.kpoint.set_grid([120,1,20])
trs.center.solver.mpidist.kptprc = 40
trs.center.solver.set_mpi_command("mpiexec -n 40")
trs.solve(energies=0.1, unit="ev")
trs.plot(filename="spin_trsm.png")
A quick remark on our result. We notice that the transmission is mainly focused on two lines in the whole Brillouin zone. The x coordinate of the lines actually correspond to the projected position of graphene Dirac points, where electrons can tunnel through.