1.3. Density of states calculation
In this section, I show how to calculate the density of states (DOS) of
bulk silicon. In principle, all I have to do is set the keyword
dos.status to true and perform a self-consistent calculation. In
practice, the DOS is calculated non-self-consistently as the number of
k-point required to converge the DOS is much higher than the number of
k-point required to converge the ground state density. To perform a DOS
calculation, copy the following input file and save it to a text file
named si_dos.txt.
info.calculationType = 'dos'
info.savepath = 'results/si_dos'
atom.element = [1 1]
atom.xyz = 10.25*[0 0 0;0.25 0.25 0.25]
domain.latvec = 10.25*[0.0 0.5 0.5;0.5 0.0 0.5;0.5 0.5 0.0]
domain.lowres = 0.5
element.species = 'si'
element.path = './Si_TM_LDA.mat'
kpoint.gridn = [10,10,10]
rho.in = 'results/si_scf'
Then pass it to RESCU and execute the program as follows
rescu -i si_dos.txt;
The DOS calculation input file is similar to the self-consistent
calculation input file. The differences are highlighted in blue.
info.calculationType is set to ’dos’ and rho.in points to
the results of the self-consistent calculation so that RESCU reads the
ground state electronic density. info.savepath is modified not to
overwrite the self-consistent results and the k-sampling is increased.
In the present case, a 10\(\times\)10\(\times\)10 grid
is sufficient since the default sampling method is the linear
tetrahedron method and the bands of silicon are relatively smooth.
1.4. Plot results
RESCU automatically plots certain results (e.g. band structures, DOS,
projected DOS). It create a MATLAB figure that can be modified (title,
axis, etc.) subsequently. To plot the density of states, use the
plot flag to point RESCU to the data.
rescu -p results/si_dos.mat results/si_dos.png;
The second input (results/si_dos.png) is optional and it determines
the name and the file format of the figure. The DOS obtained from the
example in section 3 is found in
Fig. 1.4.1.
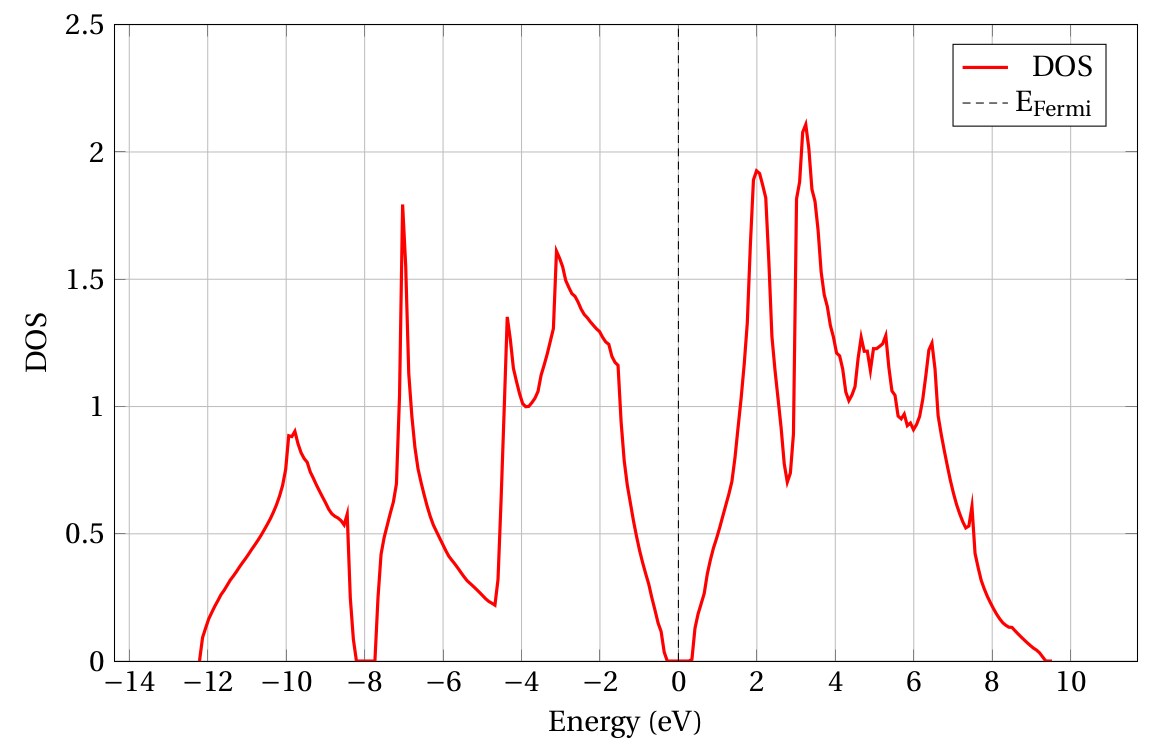
Fig. 1.4.1 DOS of bulk silicon