Local density of states tutorial
Requirements
Software components
nanotools
RESCU+
Pseudopotentials
I will need the following pseudopotentials.
C_LDA_TM_DZP.mat
Let’s copy them from the pseudo database to the current working directory and export RESCUPLUS_PSEUDO=$PWD.
References
Briefing
In this tutorial, I show how to calculate the local density of states of graphene near the Dirac point.
Setup
Create the Python script etot.py with the following content (see the tutorial on TotalEnergy)
from nanotools import Atoms, Cell, System, TotalEnergy
import numpy as np
a = 2.46 # lattice constant (ang)
avec = [
[.5*a, np.sqrt(3)/2*a, 0.],
[.5*a,-np.sqrt(3)/2*a, 0.],
[0.,0.,10.583544213400000],
]
cell = Cell(avec=avec, resolution=0.12)
fxyz = [[2/3, 1/3, 0.5],[1/3, 2/3, 0.5]]
atoms = Atoms(fractional_positions=fxyz, formula="CC")
sys = System(cell=cell, atoms=atoms)
sys.kpoint.set_grid([15,15,1])
calc = TotalEnergy(sys)
calc.solve()
and ldos.py
from nanotools import LocalDOS
import numpy as np
from nanotools.utils import ureg
calc = LocalDOS.from_totalenergy("nano_scf_out.json")
calc.system.kpoint.set_grid([30,30,1], shift = [.5, .5, 0.])
calc.system.pop.set_type("tm")
calc.ldos.energies = [(calc.energy.efermi.m - 0.01)] * ureg.electron_volt
calc.solve()
calc.plot_ldos(filename="ldos.png", show=True)
Explanations
Here is a high level view of the calculation workflow:
Initialize the LocalDOS calculator from a ground state calculation (
TotalEnergy).Perform a local density of states calculation (non-self-consistently) on a finer k-point grid and compute the local density of states.
Initialize the LocalDOS calculator
Upon completing the total energy calculation, the results are saved in nano_scf_out.json.
I will initialize a LocalDOS calculator using the class method from_totalenergy.
calc = LocalDOS.from_totalenergy("nano_scf_out.json")
calc.system.kpoint.set_grid([30,30,1], shift = [.5, .5, 0.])
calc.system.pop.set_type("tm")
calc.ldos.energies = [(calc.energy.efermi.m - 0.01)] * ureg.electron_volt
The method from_totalenergy will copy the system information and initialize the LocalDOS calculator.
I then increase the k-sampling resetting the kpoint grid with set_grid and change the population function to tm which stands for tetrahedron method.
The ldos parameters are given in the ldos attribute.
In particular, I can set the energies at which compute the local DOS setting ldos.energies.
Here I compute the local DOS 0.01 eV under the Fermi level.
Local density of states calculation
RESCU+’s solvers are invoked calling the solve method
calc.solve()
The method writes all parameters to a JSON file, then calls the relevant (Fortran) program, then loads the data back into the calculator.
The output of rescuplus goes to nano_ldos_out.h5 and nano_ldos_out.json.
As other real space data, the local DOSes are stored in nano_ldos_out.h5 similarly with density, i.e. under /ldos/#/xxx where # is an index running over all energy points and xxx is total, spin-up, spin-down, spin-x, spin-y, spin-z depending on spin.
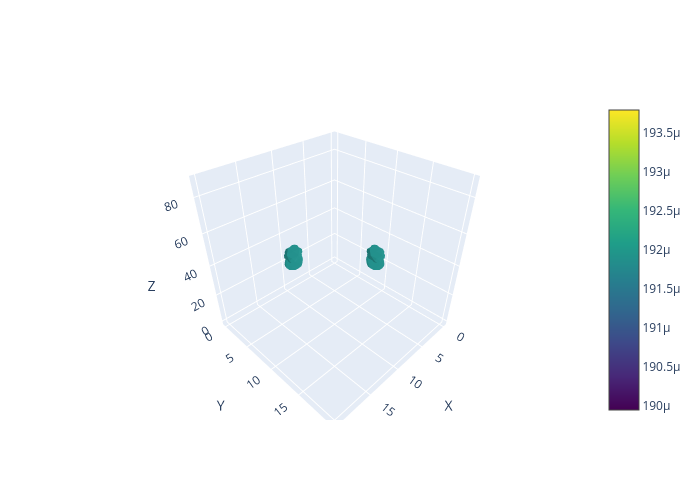
To plot and visualize the LDOS isosurface, add the following code at the end of your Python script:
# Plot with the default value
calc.plot_ldos(filename="ldos.png", show=True)
After, you will get a picture similar to this:
To plot the LDOS isosurface with a specified density level:
# Plot with a specific value
calc.plot_ldos(value=0.5, filename="ldos.png", show=True)