9.1. Example: DOS of nickel
Let’s assume that we have accomplished a self-consistent calculation of
the face-centered cubic nickel solid and that the results are in
./results/ni_lcao_scf.h5 and ./results/ni_lcao_scf.mat. One may
compute the DOS of FCC nickel using the following input script.
info.calculationType = 'dos'
info.savepath = './results/ni_lcao_dos';
kpoint.gridn = [22,22,22];
kpoint.sampling = 'tetrahedron'
rho.in = './results/ni_lcao_scf';
dos.resolution = 1e-4;
Information such as structure, pseudopotentials, functional, etc. are
read from rho.in. Here are some keywords which typically appear in a
DOS calculation input file.
info.calculationTypedetermines the nature of the calculation. If it is set todos, the eigenvalues are calculated at all non-degenerate k-points and the DOS is calculated;kpoint.gridnthis keyword determines the fineness of the reciprocal space grid (k-point grid). It corresponds to the number of points along each reciprocal lattice vectors. Note that the accuracy is more correlated with the number of irreducible k-points than the total number of k-points. In the present example, the [22,22,22] k-grid accuracy is roughly equivalent to the [35,35,35] k-grid accuracy because the latter is more redundant than the former;kpoint.samplingthis keyword determines the DOS shape functions. For example, if the keyword is set togauss, then every state is smeared using a Gaussian function. In the present example, keyword is set totetrahedron, such that the DOS is calculated using linear tetrahedral elements;rho.inpath the the result containing the self-consistent (spin) density;dos.resolutionsets the energy density for the DOS calculation. The default value is 10\(^{-3}\).
The program is run as usual. For example, if the above file is saved to
ni_lcao_dos, then we run it as follows.
rescu -i ni_lcao_dos
The results are found in ./results/ni_lcao_dos.mat. We can plot them
typing
rescu -p ./results/ni_lcao_dos.mat
which give the plot shown in Fig. 9.1.1. You may also find the DOS data
in ./results/DensityOfStates.txt.
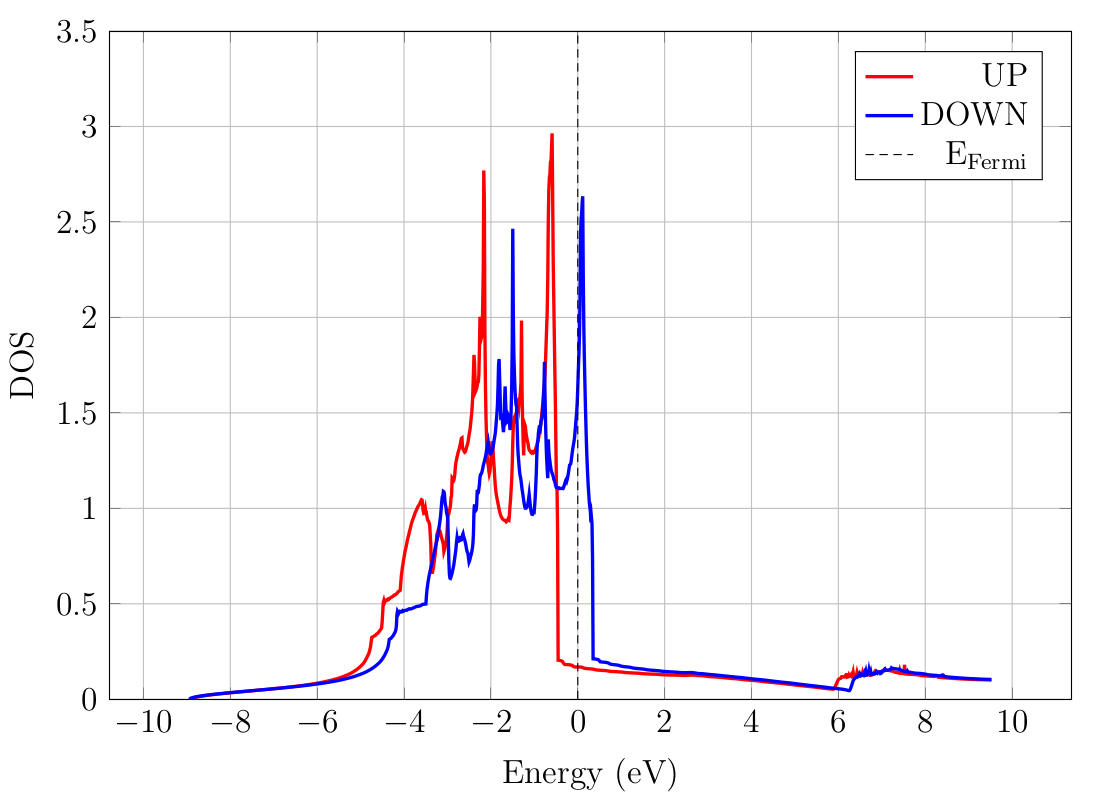
Fig. 9.1.1 DOS of FCC nickel. The calculation is done using a double-zeta polarized atomic orbital basis.