23. Electron \(\mathbf{k} \cdot \mathbf{p}\) simulation of Jackiw–Rebbi states
23.1. Requirements
23.1.1. Software components
QTCAD
Gmsh
23.1.2. Geometry file
qtcad/examples/tutorials/meshes/jackiw_rebbi_1d.geo
23.1.3. Python script
qtcad/examples/tutorials/electron_kp_jackiw_rebbi.py
23.1.4. References
23.2. Briefing
The purpose of this tutorial is to show how to use QTCAD’s general electron
\(\mathbf{k} \cdot \mathbf{p}\) module in a typical workflow.
The ElectronKPModel class allows
users to construct custom \(n\)-band Hamiltonians of the form
where \(V_{\mathrm{conf}}(\mathbf{r})\) is the confinement potential,
\(\mathbb{I}\) is the \(n \times n\) identity matrix, \(k_1 = 1\) is a
bookkeeping convention, and \(Q_{ij}\) are \(n \times n\) Hamiltonian
coefficients. In particular,
ElectronKPModel allows users to
specify the matrices \(Q_{ij}\) that multiply powers of \(k_x\), \(k_y\),
and \(k_z\) up to quadratic order. These matrices can be specified directly or
derived from Material or
Device attributes.
As a concrete example, we solve the one-dimensional Jackiw–Rebbi problem [JR76]. This example is small enough to run very quickly, even on a laptop, yet it still illustrates key \(\mathbf{k} \cdot \mathbf{p}\) ingredients that are also important in larger multiband models: a band matrix, a named material parameter, a position-dependent coefficient, and a first-order term in momentum.
23.2.1. Theory
Topologically protected bound states are localized eigenstates that appear where two gapped bulk regions with different topology meet. In the simplest one-dimensional case, this topology is set by the sign of the Dirac mass (the parameter whose magnitude sets half the bulk energy gap). While a smooth, local perturbation can deform the wavefunction and shift its energy, it cannot eliminate the bound state unless the energy gap closes or the interface between the two distinct phases is removed.
The Jackiw–Rebbi model [JR76] is the simplest continuum example of this idea. It is a one-dimensional Dirac Hamiltonian,
where \(\sigma_x\) and \(\sigma_z\) are Pauli matrices, \(\hbar v\) is a velocity parameter, and \(M(x)\) is a position-dependent (Dirac) mass term. The Hamiltonian in Eq. (23.2.2) is a special case of the general electron \(\mathbf{k} \cdot \mathbf{p}\) Hamiltonian in Eq. (23.2.1), with \(Q_{11} = M(x)\sigma_z\) and \(Q_{1x} = \hbar v\sigma_x\).
For a uniform mass, the bulk spectrum is
Equation (23.2.3) shows that a uniform mass opens an energy gap of size \(2|M|\) between the positive- and negative-energy bands. Thus, \(|M|\) controls the size of the gap, while the sign of \(M\) labels two distinct gapped phases. To continuously deform one phase into the other, the mass parameter must pass through \(M=0\), where the bulk gap closes. If this change occurs as a function of position, then the gap closing is localized at the point where \(M(x)=0\). This point is a domain wall, and the one-dimensional Dirac equation supports an exponentially localized zero-energy state there. The same mechanism underlies the domain-wall solitons of the Su–Schrieffer–Heeger chain [SSH79].
In this tutorial, \(M(x)\) is taken to be positive on the left side of the device and negative on the right side. The mass profile is
with \(M_\mathrm{L}>0\) and \(M_\mathrm{R}<0\). The resulting sign change at \(x=0\) acts as a single topological domain wall. The Jackiw–Rebbi mode is localized at this interface and appears as a near-zero-energy eigenstate of the finite numerical problem.
For the Hamiltonian in Eq. (23.2.2) and the mass profile in Eq. (23.2.4), the zero-energy Schrödinger equation can be reduced to [JR76, She17]
For this mass configuration, the normalizable solution of Eq. (23.2.5) is proportional to the \(\sigma_y\) eigenstate with eigenvalue \(-1\),
multiplied by an exponential envelope that decays away from the interface,
Both exponentials decay away from \(x=0\) because \(M_\mathrm{L}>0\) and \(M_\mathrm{R}<0\).
23.2.2. Overview
In this tutorial, we will numerically compute the bound zero-energy state and compare our result to the analytic solution.
The tutorial focuses on the following electron \(\mathbf{k} \cdot \mathbf{p}\) steps:
defining a two-band model,
adding an
ElectronKPParameterto this two-band model,adding a first-order linear-in-\(k_x\) term to this two-band model, and
solving the resulting model with the standard Schrödinger
Solver.
23.3. Mesh generation
The mesh for this tutorial is generated from the geometry file
qtcad/examples/tutorials/meshes/jackiw_rebbi_1d.geo. This geometry contains
two physical curves:
left_trivial, where \(M(x) = M_L > 0\),right_topological, where \(M(x) = M_R <0\).
It also contains two insulating point boundaries, left_bnd and right_bnd. To
regenerate the mesh file manually, run Gmsh from the mesh directory:
gmsh jackiw_rebbi_1d.geo
This produces jackiw_rebbi_1d.msh.
23.4. Header
We start by importing standard Python libraries alongside QTCAD modules used to import
a Mesh, define a
Device, set the electron
\(\mathbf{k} \cdot \mathbf{p}\) model, and solve the
Schrödinger equation.
import pathlib
import numpy as np
from numpy.typing import NDArray
from matplotlib import pyplot as plt
from qtcad.device import constants as ct
from qtcad.device import materials as mt
from qtcad.device.mesh1d import Mesh
from qtcad.device import Device
from qtcad.device.analysis import integrate
from qtcad.device.electron_kp import ElectronKPModel
from qtcad.device.electron_kp import ElectronKPParameter
from qtcad.device.pauli import sigma_vec
from qtcad.device.schrodinger import Solver
from qtcad.device.schrodinger import SolverParams
The important new imports are
ElectronKPModel and
ElectronKPParameter.
ElectronKPModel stores the
Hamiltonian coefficients for the electron \(\mathbf{k} \cdot \mathbf{p}\) model [see
Eq. (23.2.1)], while
ElectronKPParameter
is used to represent a coefficient that should be resolved by name on the
Device or on the
Material objects assigned to its regions.
23.5. Setup
We next define paths and the physical parameters of the model.
23.5.1. File paths
We start with defining the path to the mesh file and the output figure that will be produced by the script.
# File paths -----------------------------------------------------------------
script_dir = pathlib.Path(__file__).parent.resolve()
path_mesh = script_dir / "meshes" / "jackiw_rebbi_1d.msh"
path_out = script_dir / "output"
path_fig = path_out / "electron_kp_jackiw_rebbi.png"
23.5.2. Model parameters
Next, we define the model parameters
# Model parameters -----------------------------------------------------------
# Units.
nm = 1e-9
meV = ct.e / 1000
# Mass-energy term.
mass_parameter_name = "jackiw_rebbi_mass"
M_L = 2.5 * meV
M_R = -1.5 * meV
# Velocity term.
hbar_v = 25.0 * meV * nm
# Plotting window.
plot_window = 50.0 * nm
# Number of states to solve the Schrödinger equation for.
num_states = 8
The string mass_parameter_name is the key that will be used to connect the material
data to the ElectronKPParameter
appearing in the model.
We also define two mass terms: M_L, which is positive, and
M_R, which is negative. The opposite signs create the
Jackiw–Rebbi interface at the point where the two material regions meet.
Finally, we define the velocity term (or \(\hbar\) times the velocity) and set the
number of states the Schrödinger Solver
computes. The plot_window parameter restricts the final figure to the region
near the interface so this spatial structure is easier to see.
23.6. Defining the electron \(\mathbf{k} \cdot \mathbf{p}\) model
The electron \(\mathbf{k} \cdot \mathbf{p}\) model has two bands because the Jackiw–Rebbi Hamiltonian acts on a two-component spinor. We first define a helper function that transforms a scalar mass value into the matrix-valued \(k\)-independent coefficient \(M(x)\sigma_z\).
# Electron k · p model -------------------------------------------------------
def mass_term_sigma_z(mass: float) -> NDArray[np.float64]:
"""Convert a scalar mass into a Jackiw–Rebbi mass term.
Args:
mass: A scalar mass corresponding to M(x) at a given point in space.
Returns:
Matrix-valued coefficient mass * sigma_z representing the second term in the
Jackiw-Rebbi Hamiltonian M(x) * sigma_z at a given point in space.
"""
return mass * sigma_vec[2]
The extra axes make the returned object a matrix in band space. This function works both when the resolved parameter is a single scalar and when it is a spatial array sampled over the mesh.
The model itself is then defined as follows.
def build_jackiw_rebbi_model() -> ElectronKPModel:
"""Build the two-band Jackiw–Rebbi electron k·p model.
The Hamiltonian is
H = hbar v k_x sigma_x + M(x) sigma_z.
The mass term M(x) is resolved from the region materials through an
ElectronKPParameter.
The velocity term (proportional to sigma_x) is constant over all space.
Returns:
Electron k·p model describing the Jackiw–Rebbi model.
"""
mass_term = ElectronKPParameter(
mass_parameter_name, # Connect the model to material parameter.
transform=mass_term_sigma_z, # Get mass term proportional to sigma_z.
)
velocity_term = hbar_v * sigma_vec[0] # Velocity term proportional to sigma_x.
model = ElectronKPModel(
bands=2,
constant=mass_term,
linear={"x": velocity_term},
)
return model
The constant argument contributes to the \(k\)-independent term. Since it
is an ElectronKPParameter,
the value is not fixed when the model is created.
Instead, the Schrödinger Solver resolves the
parameter named mass_parameter_name on each region material and applies the
transformation mass_term_sigma_z to obtain the coefficient that enters the
Hamiltonian.
The linear argument encodes the first-order term \(\hbar v k_x\sigma_x\). The
direction key "x" is a shorthand for the ordered electron
\(\mathbf{k} \cdot \mathbf{p}\) coefficient ("1", "x"), which multiplies
\(k_x\). In this case, the linear coefficient is spatially uniform and can be passed
directly as a NumPy array.
Note
The constant and linear constructor arguments used above are
convenient shorthands for the \(Q_{11}\) and \(Q_{1j}\) matrices,
\(j \in \{x, y, z\}\). The quadratic argument may be used when explicit
ordered pairs are needed. It accepts a dictionary whose keys are pairs drawn from
("1", "x", "y", "z") and whose values are scalars, band matrices, spatial
arrays, position-dependent functions, or
ElectronKPParameter objects.
23.7. Assigning electron \(\mathbf{k} \cdot \mathbf{p}\) parameters to materials
Here, we create custom materials for the two different regions of the
Device.
Each Material acts as a convenient container
for region-specific data.
The Jackiw–Rebbi mass itself is supplied through the electron
\(\mathbf{k} \cdot \mathbf{p}\) parameter dictionary on each material.
The entries of this dictionary are set via the
set_electron_kp_param
method.
# Region materials -----------------------------------------------------------
def build_region_materials() -> tuple[mt.Material, mt.Material]:
"""Create materials carrying the electron k·p mass parameter.
Returns:
Materials representing the two different regions of the device.
Both materials possess a k·p parameter, labelled `"jackiw_rebbi_mass"`,
which is set via `set_electron_kp_param`.
"""
left_trivial = mt.Material({"name": "Left"})
left_trivial.set_electron_kp_param(
mass_parameter_name,
M_L,
)
right_topological = mt.Material({"name": "Right"})
right_topological.set_electron_kp_param(
mass_parameter_name,
M_R,
)
return left_trivial, right_topological
The parameter name used here is the same name used in
ElectronKPParameter.
This is what links the Hamiltonian definition to the
Material data.
23.8. Building the device
The device setup follows the usual QTCAD pattern: load a
Mesh, set the
Material of each region using the
new_region method, and add boundary
conditions (in this case, insulator boundaries at the two endpoints) using the
new_insulator method.
# Device setup ---------------------------------------------------------------
def build_device() -> Device:
"""Load the mesh, assign regions, and attach the electron k·p model.
Returns:
Device containing the Jackiw–Rebbi mesh, material regions, boundary
conditions, and Jackiw–Rebbi k·p model.
"""
# Create the materials.
left_trivial, right_topological = build_region_materials()
# Create the device.
mesh = Mesh(nm, str(path_mesh))
dvc = Device(mesh, conf_carriers="e")
## Set the regions.
dvc.new_region("left_trivial", left_trivial)
dvc.new_region("right_topological", right_topological)
## Set the boundary conditions.
dvc.new_insulator("left_bnd")
dvc.new_insulator("right_bnd")
## Set the k·p model.
jackiw_rebbi_model = build_jackiw_rebbi_model()
dvc.set_electron_kp_model(jackiw_rebbi_model)
return dvc
The calls to new_insulator impose
perfect-insulator boundary conditions for the Schrödinger
Solver. This means that the wavefunctions
are forced to vanish at left_bnd and right_bnd (see
Boundary conditions for the general boundary-condition
discussion). Because the zero mode is exponentially localized near \(x=0\), and
the endpoints of the mesh are placed far from the interface, we do not expect this
choice of boundary condition to appreciably influence the properties of the bound
state.
The electron \(\mathbf{k} \cdot \mathbf{p}\)-specific step is the last one which
involves attaching the
ElectronKPModel using the
set_electron_kp_model method.
Once this model is attached, the standard Schrödinger
Solver assembles the Hamiltonian from the
electron \(\mathbf{k} \cdot \mathbf{p}\) coefficients instead of from the usual
single-band effective-mass tensor.
23.9. Solving the Schrödinger equation
The next function sets up the Schrödinger
Solver, as in previous tutorials (see e.g.
Schrödinger equation for a quantum dot) and solves the Schrödinger
equation.
# Schrodinger solve ----------------------------------------------------------
def solve_schrodinger(dvc: Device) -> None:
"""Solve the electron k·p Hamiltonian with the Schrödinger solver.
Args:
dvc: Device whose electron k·p Hamiltonian is to be solved.
"""
params = SolverParams()
params.num_states = num_states
params.tol = 1e-10
params.verbose = False
solver = Solver(dvc, solver_params=params)
solver.solve()
This example has no electrostatic confinement potential. The full Hamiltonian is given
by Eq. (23.2.2).
For this reason, the Solver will warn that
the confinement potential is zero everywhere. In this tutorial that warning is expected.
23.10. Accessing the resolved mass profile
The following helper resolves the model on the device using the
resolve_device_coefficients
method and selects the constant coefficient matrix by passing the canonical pair
("1", "1") to the
get_coefficients_for_pair
method. The resolved constant coefficient is \(Q_{11} = M(x)\sigma_z\). Using the
properties of \(\sigma_z\), the mass \(M(x)\) can be obtained via
\((Q_{11,00}-Q_{11,11})/2\). We then output \(M(x)\) over the global nodes of
the device mesh.
# Resolved mass profile ------------------------------------------------------
def mass_profile_from_model(dvc: Device) -> NDArray[np.float64]:
"""Return the resolved Jackiw–Rebbi mass profile on global mesh nodes.
Args:
dvc: Device with an attached Jackiw–Rebbi electron k·p model.
Returns:
Jackiw–Rebbi mass profile on global nodes of the mesh over which dvc is
defined.
"""
# Get all coefficients resolved over the device.
resolved = dvc.electron_kp_model.resolve_device_coefficients(dvc)
# Get the constant term.
constant_term = resolved.get_coefficients_for_pair(("1", "1"))
# Convert to global nodes.
constant_term_nodes = dvc.mesh.toglobal(constant_term[0])
# Get the M(x) parameter.
mass_term = 0.5 * (
constant_term_nodes[..., 0, 0] - constant_term_nodes[..., 1, 1]
)
mass_term = np.real(mass_term)
return mass_term
23.11. Analytic bound state
The Briefing section presented the analytic zero mode for the sign-changing mass profile. We now define a helper function that evaluates Eq. (23.2.6) on the global nodes of the same mesh used by the numerical problem.
# Analytic bound state -------------------------------------------------------
def analytic_bound_state(
mesh: Mesh,
) -> NDArray[np.complex128]:
"""Return the analytic Jackiw–Rebbi zero mode on global mesh nodes.
Args:
mesh: Mesh over which the analytic state is defined.
Returns:
Normalized two-component analytic bound-state wavefunction sampled on the
mesh global nodes.
"""
x = mesh.glob_nodes[:, 0]
envelope = np.where(
x < 0.0,
np.exp(M_L * x / hbar_v),
np.exp(M_R * x / hbar_v),
)
# The zero-energy mode is proportional to the sigma_y eigenstate.
spinor = np.array([1.0, -1.0j], dtype=complex) / np.sqrt(2.0)
wavefunction = envelope[:, np.newaxis] * spinor[np.newaxis, :]
# Apply the analytic normalization factor.
norm = np.sqrt(
-2 * M_L * M_R
/ ((M_L - M_R) * hbar_v)
)
return wavefunction * norm
Next, we define compare the numerical and analytic states. The
band weights of the numerical state are obtained from
band_weight, while the analytic
weights are obtained by integrating the component densities directly with
band_weights_from_wavefunction.
Equation (23.2.6) predicts that the two band components have
the same magnitude and differ by a phase of \(-\pi/2\). We check this relative
phase with relative_band_phase, which computes the phase of the integrated overlap
between the two band components.
def band_weights_from_wavefunction(
mesh: Mesh,
wavefunction: NDArray[np.complex128],
) -> NDArray[np.float64]:
"""Integrate the band weights of a two-component wavefunction.
Args:
mesh: Mesh over which the wavefunction is defined.
wavefunction: Two-component wavefunction sampled on the global nodes of mesh.
Returns:
Normalized probability weight carried by each band component.
"""
component_densities = np.abs(wavefunction) ** 2
weights = np.array(
[
integrate(mesh, component_densities[:, band_index])
for band_index in range(component_densities.shape[-1])
],
)
return weights / np.sum(weights)
def relative_band_phase(
mesh: Mesh,
wavefunction: NDArray[np.complex128],
) -> float:
"""Return the relative phase between the two band components.
Args:
mesh: Mesh over which the wavefunction is defined.
wavefunction: Two-component wavefunction sampled on the global nodes of mesh.
Returns:
Phase angle, in radians, of the integrated interband coherence.
"""
coherence = integrate(mesh, np.conj(wavefunction[:, 0]) * wavefunction[:, 1])
return np.angle(coherence)
23.12. Visualizing results
We also create a function to plot the mass profile and compare the numerical near-zero-state probability density to the analytic density.
# Plotting -------------------------------------------------------------------
def plot_near_zero_states(
x: NDArray[np.float64],
mass_profile: NDArray[np.float64],
num_wavefunction: NDArray[np.complex128],
an_wavefunction: NDArray[np.complex128],
) -> None:
"""Plot the mass profile and compare numerical and analytic densities.
Args:
x: Mesh-node coordinates sorted by increasing x coordinate.
mass_profile: Jackiw–Rebbi mass profile sampled on the sorted nodes.
num_wavefunctions: Multiband eigenfunctions sorted on the same nodes.
an_wavefunction: Analytic bound-state wavefunction sampled on x.
"""
# Get densities associated to wavefunctions.
num_density = np.sum(np.abs(num_wavefunction) ** 2, axis=-1)
analytic_density = np.sum(np.abs(an_wavefunction) ** 2, axis=-1)
# Scale the positions.
x_nm = x / nm
# Create the figure.
fig, axes = plt.subplots(2, 1, figsize=(7.0, 6.0), sharex=True)
## Mass profile plot.
axes[0].plot(x_nm, mass_profile / meV, color="black", linewidth=2.0)
axes[0].axhline(0.0, color="0.5", linewidth=0.8)
axes[0].axvline(0.0, color="0.5", linestyle="--")
axes[0].set_ylabel("M(x) [meV]")
axes[0].set_title("Jackiw–Rebbi mass profile")
axes[0].grid(alpha=0.25)
## Densities plot.
num_style = {"linewidth": 3.0, "linestyle": "-"}
axes[1].plot(
x_nm,
num_density * nm,
label="numerical zero mode",
**num_style,
)
an_style = {"linewidth": 2.0, "linestyle": ":", "color": "black"}
axes[1].plot(
x_nm,
analytic_density * nm,
label="analytic zero mode",
**an_style
)
axes[1].axvline(0.0, color="0.5", linestyle="--")
axes[1].set_xlabel("x [nm]")
axes[1].set_ylabel("Probability density [1/nm]")
axes[1].set_title("Near-zero-state density")
axes[1].grid(alpha=0.25)
axes[1].legend()
axes[1].set_xlim(-plot_window / nm, plot_window / nm)
fig.tight_layout()
# Save the figure.
path_out.mkdir(parents=True, exist_ok=True)
fig.savefig(path_fig, dpi=200, bbox_inches="tight")
plt.close(fig)
23.13. Analyzing the near-zero state
23.13.1. Simulation
Now that all the functions have been defined, we start with the analysis of the
near-zero-energy state of the Jackiw–Rebbi model.
The first step is set up the Device and solve the
Schrödinger equation.
# Analysis -------------------------------------------------------------------
if __name__ == "__main__":
# Create the device.
dvc = build_device()
# Solve the Schrödinger equation.
solve_schrodinger(dvc)
# Print the k·p model attached to the device.
dvc.electron_kp_model.print()
We also print the
ElectronKPModel. This prints the
coefficients associated to each term in the Hamiltonian of
Eq. (23.2.2).
ElectronKPModel
bands: 2
parameters: ('jackiw_rebbi_mass',)
canonical_terms:
[1] ('1', '1'):
ElectronKPParameter(name='jackiw_rebbi_mass', device_attr=None, material_attr=None, default=<object object at 0x135091b50>, transform='mass_term_sigma_z')
[2] ('1', 'x'):
[[0.000000e+00+0.j, 4.005442e-30+0.j],
[4.005442e-30+0.j, 0.000000e+00+0.j]]
Above is the output of print.
The model has two bands, so the eigenfunctions stored in dvc.eigenfunctions
are three-dimensional arrays. The first axis indexes the global mesh nodes, the second
axis indexes the eigenstates, and the third axis indexes the two band components.
The printed canonical_terms list shows the terms that enter the Hamiltonian.
The first coefficient, [1], is attached to the k-independent term
('1', '1'). Its value is not a fixed array in the model definition. Instead,
it is represented by an
ElectronKPParameter
named 'jackiw_rebbi_mass_energy'. The Schrödinger
Solver resolves the parameter from the
material assigned to each region and then transforms it with mass_term_sigma_z to
obtain the corresponding matrix-valued coefficient.
The second coefficient, [2], is attached to the term linear in \(k_x\),
represented by ('1', 'x'). Unlike the mass term, this coefficient is given as a
NumPy array of shape (bands, bands), so it is spatially uniform across the device.
Next, we extract the numerical state with energy closest to zero. The
Solver stores the eigenenergies in
dvc.energies and the multiband eigenfunctions in dvc.eigenfunctions.
We also compute the band weights of the numerical eigenstates with the
band_weight method. For comparison, we
evaluate the analytic bound state on the same global mesh nodes and compute its band
weights directly from the analytic wavefunction.
# Energies of the states.
energies_mev = dvc.energies / meV
# Find the near-zero energy state.
near_zero_index = np.argsort(np.abs(energies_mev))[0]
# Weight of the eigenstates projected onto each band.
band_weights = dvc.band_weight()
# Numerical bound state on global nodes.
num_wavefunction = dvc.eigenfunctions[:, near_zero_index, :]
# Compute the analytic bound state on the global nodes.
an_wavefunction = analytic_bound_state(dvc.mesh)
# Weight of the analytic wavefunction projected onto each band.
analytic_band_weights = band_weights_from_wavefunction(
dvc.mesh,
an_wavefunction,
)
23.13.2. Comparison
With the numerical and analytic states sampled on the same global mesh nodes, we can now compare scalar quantities before turning to the spatial density plot.
We first print the selected state index and its energy.
print(f"\nState {near_zero_index}")
print("Energy [meV]:")
print(energies_mev[near_zero_index])
The selected state has energy of order \(10^{-15}\,\mathrm{meV}\), confirming that it is numerically very close to the expected zero mode.
State 4
Energy [meV]:
-5.667663519630012e-15
We then compare the numerical and analytic states more directly. In particular, we check the weight carried by each band component and the relative phase between the two components.
print("\nAnalytic comparison")
print("Numerical band weights:")
print(band_weights[near_zero_index])
print("Analytic band weights:")
print(analytic_band_weights)
print("Numerical relative phase [rad]:")
print(relative_band_phase(dvc.mesh, num_wavefunction))
print("Analytic relative phase [rad]:")
print(relative_band_phase(dvc.mesh, an_wavefunction))
Both agree with the analytic prediction. The near-zero state has equal weight in the two bands and a relative phase of \(-\pi/2\).
Analytic comparison
Numerical band weights:
[0.5000001 0.4999999]
Analytic band weights:
[0.5 0.5]
Numerical relative phase [rad]:
-1.5707963267902991
Analytic relative phase [rad]:
-1.5707963267948966
Finally, we prepare the data for visualization. The global nodes of the
Mesh are not guaranteed to be ordered by position,
so we first sort them by their x coordinate. We then apply the same ordering to the
numerical wavefunction, the analytic wavefunction, and the resolved mass profile before
plotting.
# Post-process data for visualization.
## Order the global nodes.
order = np.argsort(dvc.mesh.glob_nodes[:, 0], kind="stable")
x = dvc.mesh.glob_nodes[order, 0]
## Order the wavefunctions
num_wavefunction = num_wavefunction[order, :]
an_wavefunction = an_wavefunction[order, :]
# Order the mass, M(x).
mass_profile = mass_profile_from_model(dvc)
mass_profile = mass_profile[order]
plot_near_zero_states(
x,
mass_profile,
num_wavefunction,
an_wavefunction,
)
The resulting plot is saved to
examples/tutorials/output/electron_kp_jackiw_rebbi.png. It shows the resolved mass
profile \(M(x)\) together with the numerical and analytic probability densities for
the near-zero state.
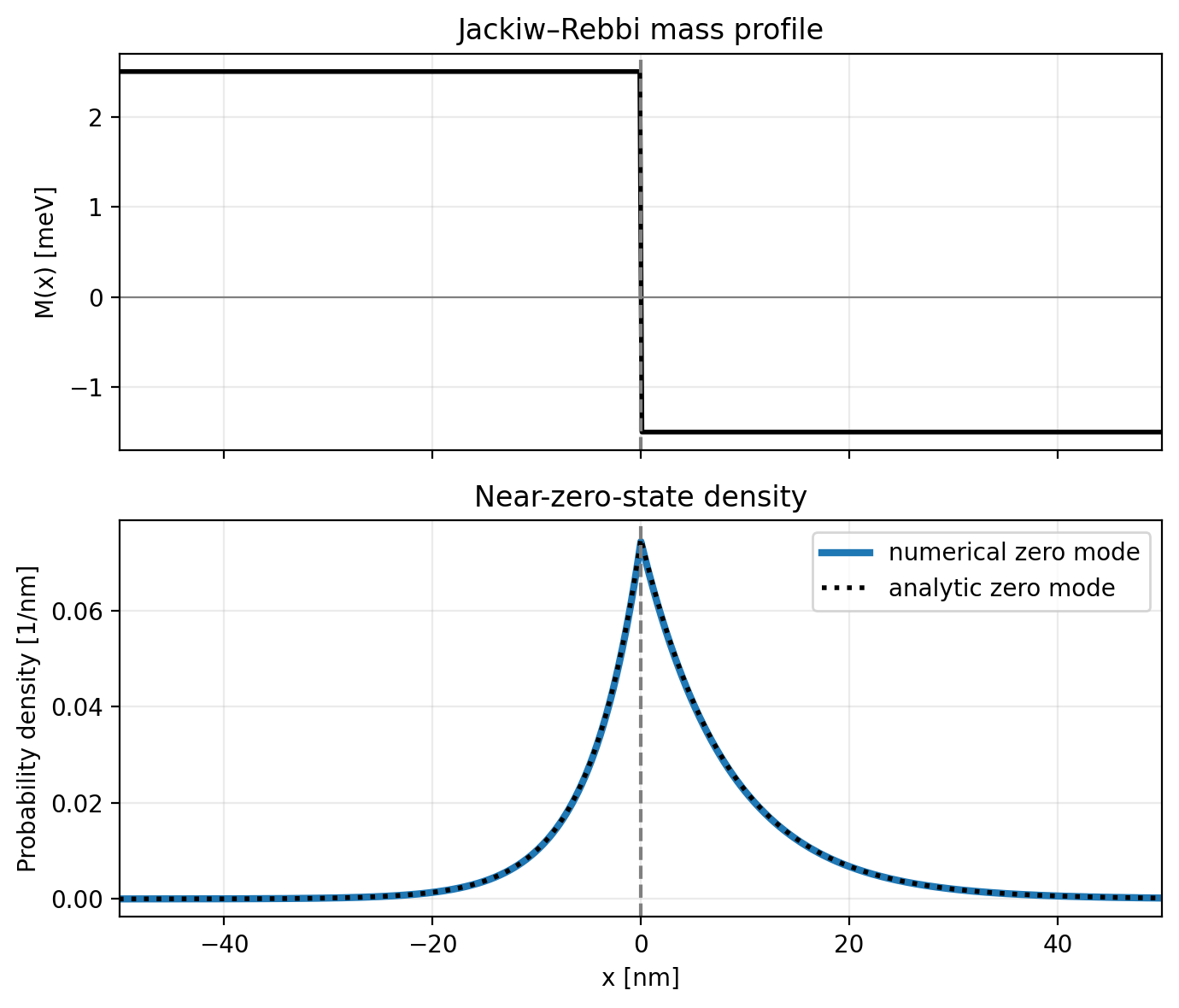
Fig. 23.13.1 Mass profile and numerical and analytic probability densities of the near-zero Jackiw–Rebbi state.
As expected, the bound state is localized at the interface where the mass term changes sign, and its numerical density overlaps with the analytic envelope from Eq. (23.2.6).
23.14. Full code
__copyright__ = "Copyright 2022-2026, Nanoacademic Technologies Inc."
import pathlib
import numpy as np
from numpy.typing import NDArray
from matplotlib import pyplot as plt
from qtcad.device import constants as ct
from qtcad.device import materials as mt
from qtcad.device.mesh1d import Mesh
from qtcad.device import Device
from qtcad.device.analysis import integrate
from qtcad.device.electron_kp import ElectronKPModel
from qtcad.device.electron_kp import ElectronKPParameter
from qtcad.device.pauli import sigma_vec
from qtcad.device.schrodinger import Solver
from qtcad.device.schrodinger import SolverParams
# File paths -----------------------------------------------------------------
script_dir = pathlib.Path(__file__).parent.resolve()
path_mesh = script_dir / "meshes" / "jackiw_rebbi_1d.msh"
path_out = script_dir / "output"
path_fig = path_out / "electron_kp_jackiw_rebbi.png"
# Model parameters -----------------------------------------------------------
# Units.
nm = 1e-9
meV = ct.e / 1000
# Mass-energy term.
mass_parameter_name = "jackiw_rebbi_mass"
M_L = 2.5 * meV
M_R = -1.5 * meV
# Velocity term.
hbar_v = 25.0 * meV * nm
# Plotting window.
plot_window = 50.0 * nm
# Number of states to solve the Schrödinger equation for.
num_states = 8
# Electron k · p model -------------------------------------------------------
def mass_term_sigma_z(mass: float) -> NDArray[np.float64]:
"""Convert a scalar mass into a Jackiw–Rebbi mass term.
Args:
mass: A scalar mass corresponding to M(x) at a given point in space.
Returns:
Matrix-valued coefficient mass * sigma_z representing the second term in the
Jackiw-Rebbi Hamiltonian M(x) * sigma_z at a given point in space.
"""
return mass * sigma_vec[2]
def build_jackiw_rebbi_model() -> ElectronKPModel:
"""Build the two-band Jackiw–Rebbi electron k·p model.
The Hamiltonian is
H = hbar v k_x sigma_x + M(x) sigma_z.
The mass term M(x) is resolved from the region materials through an
ElectronKPParameter.
The velocity term (proportional to sigma_x) is constant over all space.
Returns:
Electron k·p model describing the Jackiw–Rebbi model.
"""
mass_term = ElectronKPParameter(
mass_parameter_name, # Connect the model to material parameter.
transform=mass_term_sigma_z, # Get mass term proportional to sigma_z.
)
velocity_term = hbar_v * sigma_vec[0] # Velocity term proportional to sigma_x.
model = ElectronKPModel(
bands=2,
constant=mass_term,
linear={"x": velocity_term},
)
return model
# Region materials -----------------------------------------------------------
def build_region_materials() -> tuple[mt.Material, mt.Material]:
"""Create materials carrying the electron k·p mass parameter.
Returns:
Materials representing the two different regions of the device.
Both materials possess a k·p parameter, labelled `"jackiw_rebbi_mass"`,
which is set via `set_electron_kp_param`.
"""
left_trivial = mt.Material({"name": "Left"})
left_trivial.set_electron_kp_param(
mass_parameter_name,
M_L,
)
right_topological = mt.Material({"name": "Right"})
right_topological.set_electron_kp_param(
mass_parameter_name,
M_R,
)
return left_trivial, right_topological
# Device setup ---------------------------------------------------------------
def build_device() -> Device:
"""Load the mesh, assign regions, and attach the electron k·p model.
Returns:
Device containing the Jackiw–Rebbi mesh, material regions, boundary
conditions, and Jackiw–Rebbi k·p model.
"""
# Create the materials.
left_trivial, right_topological = build_region_materials()
# Create the device.
mesh = Mesh(nm, str(path_mesh))
dvc = Device(mesh, conf_carriers="e")
## Set the regions.
dvc.new_region("left_trivial", left_trivial)
dvc.new_region("right_topological", right_topological)
## Set the boundary conditions.
dvc.new_insulator("left_bnd")
dvc.new_insulator("right_bnd")
## Set the k·p model.
jackiw_rebbi_model = build_jackiw_rebbi_model()
dvc.set_electron_kp_model(jackiw_rebbi_model)
return dvc
# Schrodinger solve ----------------------------------------------------------
def solve_schrodinger(dvc: Device) -> None:
"""Solve the electron k·p Hamiltonian with the Schrödinger solver.
Args:
dvc: Device whose electron k·p Hamiltonian is to be solved.
"""
params = SolverParams()
params.num_states = num_states
params.tol = 1e-10
params.verbose = False
solver = Solver(dvc, solver_params=params)
solver.solve()
# Resolved mass profile ------------------------------------------------------
def mass_profile_from_model(dvc: Device) -> NDArray[np.float64]:
"""Return the resolved Jackiw–Rebbi mass profile on global mesh nodes.
Args:
dvc: Device with an attached Jackiw–Rebbi electron k·p model.
Returns:
Jackiw–Rebbi mass profile on global nodes of the mesh over which dvc is
defined.
"""
# Get all coefficients resolved over the device.
resolved = dvc.electron_kp_model.resolve_device_coefficients(dvc)
# Get the constant term.
constant_term = resolved.get_coefficients_for_pair(("1", "1"))
# Convert to global nodes.
constant_term_nodes = dvc.mesh.toglobal(constant_term[0])
# Get the M(x) parameter.
mass_term = 0.5 * (constant_term_nodes[..., 0, 0] - constant_term_nodes[..., 1, 1])
mass_term = np.real(mass_term)
return mass_term
# Analytic bound state -------------------------------------------------------
def analytic_bound_state(
mesh: Mesh,
) -> NDArray[np.complex128]:
"""Return the analytic Jackiw–Rebbi zero mode on global mesh nodes.
Args:
mesh: Mesh over which the analytic state is defined.
Returns:
Normalized two-component analytic bound-state wavefunction sampled on the
mesh global nodes.
"""
x = mesh.glob_nodes[:, 0]
envelope = np.where(
x < 0.0,
np.exp(M_L * x / hbar_v),
np.exp(M_R * x / hbar_v),
)
# The zero-energy mode is proportional to the sigma_y eigenstate.
spinor = np.array([1.0, -1.0j], dtype=complex) / np.sqrt(2.0)
wavefunction = envelope[:, np.newaxis] * spinor[np.newaxis, :]
# Apply the analytic normalization factor.
norm = np.sqrt(-2 * M_L * M_R / ((M_L - M_R) * hbar_v))
return wavefunction * norm
def band_weights_from_wavefunction(
mesh: Mesh,
wavefunction: NDArray[np.complex128],
) -> NDArray[np.float64]:
"""Integrate the band weights of a two-component wavefunction.
Args:
mesh: Mesh over which the wavefunction is defined.
wavefunction: Two-component wavefunction sampled on the global nodes of mesh.
Returns:
Normalized probability weight carried by each band component.
"""
component_densities = np.abs(wavefunction) ** 2
weights = np.array(
[
integrate(mesh, component_densities[:, band_index])
for band_index in range(component_densities.shape[-1])
],
)
return weights / np.sum(weights)
def relative_band_phase(
mesh: Mesh,
wavefunction: NDArray[np.complex128],
) -> float:
"""Return the relative phase between the two band components.
Args:
mesh: Mesh over which the wavefunction is defined.
wavefunction: Two-component wavefunction sampled on the global nodes of mesh.
Returns:
Phase angle, in radians, of the integrated interband coherence.
"""
coherence = integrate(mesh, np.conj(wavefunction[:, 0]) * wavefunction[:, 1])
return np.angle(coherence)
# Plotting -------------------------------------------------------------------
def plot_near_zero_states(
x: NDArray[np.float64],
mass_profile: NDArray[np.float64],
num_wavefunction: NDArray[np.complex128],
an_wavefunction: NDArray[np.complex128],
) -> None:
"""Plot the mass profile and compare numerical and analytic densities.
Args:
x: Mesh-node coordinates sorted by increasing x coordinate.
mass_profile: Jackiw–Rebbi mass profile sampled on the sorted nodes.
num_wavefunctions: Multiband eigenfunctions sorted on the same nodes.
an_wavefunction: Analytic bound-state wavefunction sampled on x.
"""
# Get densities associated to wavefunctions.
num_density = np.sum(np.abs(num_wavefunction) ** 2, axis=-1)
analytic_density = np.sum(np.abs(an_wavefunction) ** 2, axis=-1)
# Scale the positions.
x_nm = x / nm
# Create the figure.
fig, axes = plt.subplots(2, 1, figsize=(7.0, 6.0), sharex=True)
## Mass profile plot.
axes[0].plot(x_nm, mass_profile / meV, color="black", linewidth=2.0)
axes[0].axhline(0.0, color="0.5", linewidth=0.8)
axes[0].axvline(0.0, color="0.5", linestyle="--")
axes[0].set_ylabel("M(x) [meV]")
axes[0].set_title("Jackiw–Rebbi mass profile")
axes[0].grid(alpha=0.25)
## Densities plot.
num_style = {"linewidth": 3.0, "linestyle": "-"}
axes[1].plot(
x_nm,
num_density * nm,
label="numerical zero mode",
**num_style,
)
an_style = {"linewidth": 2.0, "linestyle": ":", "color": "black"}
axes[1].plot(x_nm, analytic_density * nm, label="analytic zero mode", **an_style)
axes[1].axvline(0.0, color="0.5", linestyle="--")
axes[1].set_xlabel("x [nm]")
axes[1].set_ylabel("Probability density [1/nm]")
axes[1].set_title("Near-zero-state density")
axes[1].grid(alpha=0.25)
axes[1].legend()
axes[1].set_xlim(-plot_window / nm, plot_window / nm)
fig.tight_layout()
# Save the figure.
path_out.mkdir(parents=True, exist_ok=True)
fig.savefig(path_fig, dpi=200, bbox_inches="tight")
plt.close(fig)
# Analysis -------------------------------------------------------------------
if __name__ == "__main__":
# Create the device.
dvc = build_device()
# Solve the Schrödinger equation.
solve_schrodinger(dvc)
# Print the k·p model attached to the device.
dvc.electron_kp_model.print()
# Energies of the states.
energies_mev = dvc.energies / meV
# Find the near-zero energy state.
near_zero_index = np.argsort(np.abs(energies_mev))[0]
# Weight of the eigenstates projected onto each band.
band_weights = dvc.band_weight()
# Numerical bound state on global nodes.
num_wavefunction = dvc.eigenfunctions[:, near_zero_index, :]
# Compute the analytic bound state on the global nodes.
an_wavefunction = analytic_bound_state(dvc.mesh)
# Weight of the analytic wavefunction projected onto each band.
analytic_band_weights = band_weights_from_wavefunction(
dvc.mesh,
an_wavefunction,
)
print(f"\nState {near_zero_index}")
print("Energy [meV]:")
print(energies_mev[near_zero_index])
print("\nAnalytic comparison")
print("Numerical band weights:")
print(band_weights[near_zero_index])
print("Analytic band weights:")
print(analytic_band_weights)
print("Numerical relative phase [rad]:")
print(relative_band_phase(dvc.mesh, num_wavefunction))
print("Analytic relative phase [rad]:")
print(relative_band_phase(dvc.mesh, an_wavefunction))
# Post-process data for visualization.
## Order the global nodes.
order = np.argsort(dvc.mesh.glob_nodes[:, 0], kind="stable")
x = dvc.mesh.glob_nodes[order, 0]
## Order the wavefunctions
num_wavefunction = num_wavefunction[order, :]
an_wavefunction = an_wavefunction[order, :]
# Order the mass, M(x).
mass_profile = mass_profile_from_model(dvc)
mass_profile = mass_profile[order]
plot_near_zero_states(
x,
mass_profile,
num_wavefunction,
an_wavefunction,
)